Автор: Денис Аветисян
В статье представлен метод стохастической тензорной свертки, позволяющий существенно сократить вычислительные затраты в квантхимических расчетах.

Стохастическая тензорная свертка (STC) обеспечивает масштабирование, сравнимое с теорией среднего поля, при сохранении высокой точности расчетов в рамках теории сцепленных кластеров.
Вычислительные методы в квантовой химии часто сталкиваются с экспоненциальным ростом затрат при работе с многоэлектронными системами. В данной работе, посвященной ‘Stochastic tensor contraction for quantum chemistry’, представлен стохастический метод свёртки тензоров, значительно снижающий вычислительную сложность таких операций. В частности, показано, что предложенный подход позволяет достичь масштабирования, сравнимого с методами среднего поля, при сохранении химической точности, и успешно применяется к теории сцепленных кластеров с учётом тройных возбуждений. Не открывает ли это путь к моделированию ещё более сложных химических систем, ранее недоступных для точных квантово-химических расчетов?
Электронная корреляция: вызов для современной химии
Точное моделирование молекулярных свойств требует учета электронной корреляции — взаимодействия между электронами, которое выходит за рамки простых приближений. В то время как методы, такие как теория Хартри-Фока, часто дают лишь базовое представление о поведении электронов, игнорируя их мгновенные взаимодействия, реальные молекулы демонстрируют гораздо более сложное поведение. Это взаимодействие, возникающее из-за кулоновского отталкивания между электронами, приводит к корреляции их движения, что существенно влияет на такие свойства, как энергия связи, геометрия молекулы и спектроскопические характеристики. Игнорирование электронной корреляции приводит к неточным результатам, особенно для систем с сильно коррелированными электронами, что делает ее критически важным аспектом в квантовой химии и материаловедении. Учет этих сложных взаимодействий является ключом к созданию надежных моделей и предсказанию свойств молекул и материалов с высокой точностью.
Традиционные методы, такие как теория Хартри-Фока, зачастую оказываются недостаточными для точного моделирования молекулярных свойств. В основе этой теории лежит упрощающее предположение о независимости электронов, игнорирующее мгновенные корреляции между ними, возникающие из-за кулоновского отталкивания. Это приводит к систематическим ошибкам в расчетах энергии и других важных характеристик молекул. В результате, для получения более реалистичных результатов необходимы более сложные подходы, учитывающие электронную корреляцию. Эти методы, хотя и более точные, требуют значительно больших вычислительных ресурсов, что ограничивает их применение к крупным молекулам и сложным системам. Следовательно, поиск эффективных и масштабируемых методов учета электронной корреляции является одной из ключевых задач современной квантовой химии.
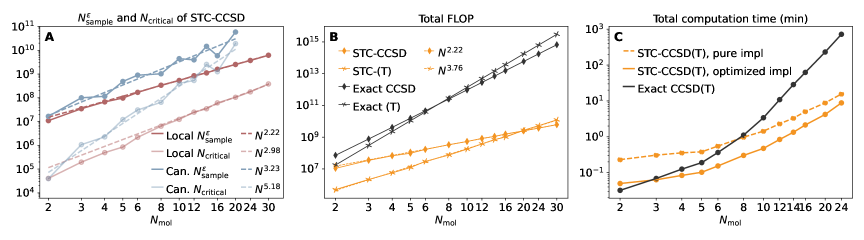
Вычислительная стоимость точного учёта электронной корреляции резко возрастает с увеличением размера исследуемой системы, что существенно ограничивает возможности моделирования. Традиционный метод, известный как CCSD(T), обладает масштабированием порядка O(N^7), где N — число базисных функций, описывающих электронную структуру. Это означает, что для удваивания размера системы, вычислительные затраты возрастают в 128 раз. Такой экспоненциальный рост препятствует применению CCSD(T) к крупным молекулам или системам с большим числом электронов, требуя разработки более эффективных алгоритмов и приближений для преодоления этого вычислительного барьера и расширения границ применимости высокоточных квантово-химических расчётов.
Тензорные свёртки: узкое место вычислений
Операция свёртки тензоров (TensorContraction) является фундаментальной для большинства расчётов в квантовой химии, поскольку она обеспечивает математическое описание электронных взаимодействий. В частности, электронная структура молекул описывается волновой функцией, которая представляется в виде многомерного тензора. Для вычисления различных свойств молекулы, таких как энергия и дипольный момент, необходимо выполнять свёртки этого тензора с другими тензорами, представляющими операторы и базисные функции. Свёртка тензоров позволяет эффективно вычислить вклад различных электронных корреляций в общую энергию и другие свойства, что критически важно для достижения высокой точности расчётов. \contraction{A}{i}{j}{B} — пример обозначения свёртки тензоров, где индексы, по которым выполняется суммирование, указываются явно.
Вычислительная сложность операции свёртки тензоров (TensorContraction) быстро становится ограничивающим фактором при моделировании крупных молекул и проведении высокоточных расчётов. Сложность данной операции растёт экспоненциально с увеличением размера системы, что обусловлено необходимостью обработки огромного количества взаимодействий между электронами. Например, для расчёта корреляционных поправок в методе теории возмущений, сложность может достигать O(N^7), где N — количество базисных функций, используемых для описания молекулы. Это означает, что даже умеренное увеличение размера молекулы может привести к многократному увеличению времени вычислений и требуемых ресурсов, делая применение точных методов непрактичным для больших систем.
Высокоточные методы квантовой химии, такие как теория сцепленных кластеров (Coupled Cluster Theory), в значительной степени зависят от эффективности алгоритмов выполнения операции свёртки тензоров (Tensor Contraction). Традиционные вычисления CCSD(T) характеризуются масштабированием сложности O(N7), где N — размер базисного набора. Это означает, что вычислительные затраты растут пропорционально N в седьмой степени, что существенно ограничивает применимость метода к большим молекулам и системам, требующим высокой точности расчётов. Эффективная реализация алгоритмов свёртки тензоров является ключевым фактором для преодоления этого ограничения и расширения возможностей высокоточных квантово-химических расчётов.
Стохастическая свёртка тензоров: новый подход к вычислениям
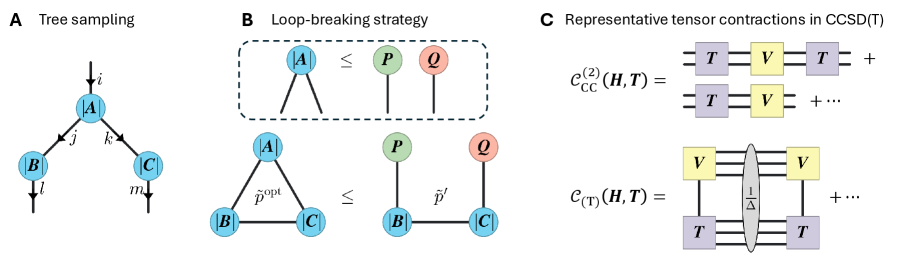
Метод Stochastic Tensor Contraction (STC) представляет собой перспективную альтернативу традиционным подходам к тензорному свёртыванию, позволяющую снизить вычислительные затраты за счёт использования стохастической выборки. Вместо вычисления всех возможных комбинаций, STC использует случайные выборки для аппроксимации результата, что особенно эффективно для систем с высокой размерностью. Это позволяет значительно уменьшить объём вычислений, сохраняя при этом приемлемую точность, и потенциально масштабировать вычисления на более крупные системы, недоступные для точных методов. Снижение вычислительной сложности достигается за счёт замены детерминированных операций на стохастические, что приводит к уменьшению требуемой памяти и времени вычислений.
Метод Stochastic Tensor Contraction (STC) развивает и расширяет принципы теории среднего поля (Mean Field Theory, MFT), предлагая более эффективный подход к аппроксимации сложных взаимодействий. В отличие от MFT, которая рассматривает среднее воздействие всех частиц на каждую отдельную частицу, STC использует стохастическую выборку для оценки вклада различных взаимодействий, что позволяет снизить вычислительную сложность. Такой подход позволяет приближенно решать задачи, требующие учета множества взаимодействий между элементами системы, сохраняя при этом приемлемую точность и масштабируемость. STC позволяет моделировать системы, в которых традиционные методы становятся непрактичными из-за экспоненциального роста вычислительных затрат с увеличением числа частиц или взаимодействий.
В рамках теории сцепленных кластеров, приближение замороженных ядер часто используется для упрощения вычислений, однако в системах с отсутствием запрещенной зоны, известных как gapless systems, это может приводить к существенным погрешностям. Данное приближение, исключая электроны внутренней оболочки из корреляционных расчетов, может недооценивать важные взаимодействия, влияющие на свойства материала. В подобных системах, где даже слабые электронные корреляции играют решающую роль, игнорирование вклада электронов внутренней оболочки может привести к неточным результатам, снижая надежность предсказаний свойств и поведения материала. Поэтому, для получения достоверных данных о gapless системах, необходимо тщательно оценивать влияние приближения замороженных ядер и, при необходимости, использовать более точные, хотя и вычислительно затратные, методы.
Сравнение с методами локальной корреляции: взгляд в будущее
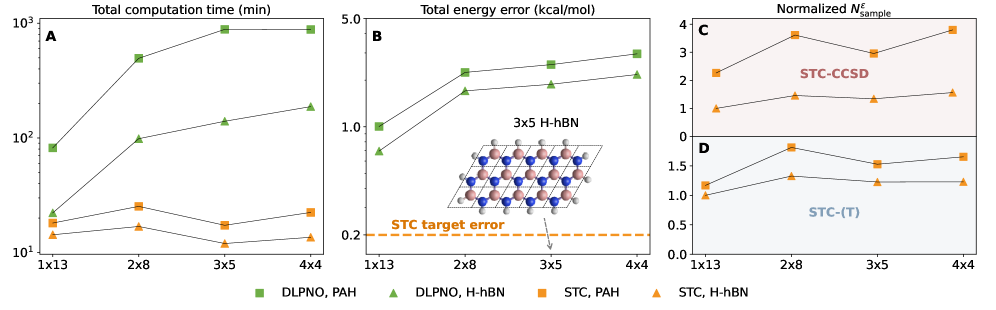
Методы локальной корреляции, такие как DomainLocalizedPairNaturalOrbitals (DLPNO), представляют собой альтернативные подходы к снижению вычислительных затрат в квантово-химических расчетах. Эти методы достигают этого путем локализации интегралов корреляции, что позволяет эффективно отбрасывать незначительные взаимодействия между удаленными областями молекулы. Вместо вычисления всех электрон-электронных взаимодействий, DLPNO фокусируется на взаимодействиях внутри определенных доменов, определенных на основе локальных орбиталей. Это существенно снижает вычислительную сложность, позволяя проводить расчеты для более крупных молекул, хотя и с потенциальной потерей точности, обусловленной усечением дальнодействующих взаимодействий.
Методы локальной корреляции, такие как DomainLocalizedPairNaturalOrbitals, хоть и эффективны в снижении вычислительных затрат, могут приводить к потере точности из-за усечения дальних взаимодействий. В стандартных методах корреляции электронная корреляция описывается за счет взаимодействия всех электронов в системе. Локальные приближения ограничивают это взаимодействие только ближайшими электронами или определенной областью пространства, что может игнорировать важные, но более слабые, корреляционные эффекты, возникающие между удаленными электронами. Это усечение приводит к систематической ошибке, особенно при расчете свойств, чувствительных к дальним взаимодействиям, таких как поляризуемость или энергия диссоциации.
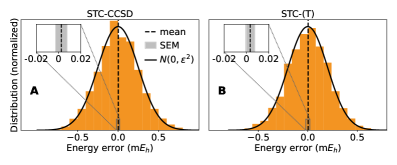
Метод стохастической тензорной контракции (StochasticTensorContraction) рассматривается в качестве перспективной альтернативы традиционным методам локальной корреляции. В частности, STC-CCSD(T) демонстрирует среднюю абсолютную ошибку (MAE) менее 0.22 ккал/моль на эталонном наборе молекул, что свидетельствует о высокой точности. Кроме того, STC-CCSD(T) обеспечивает ускорение вычислений до 32x по сравнению с методом DLPNO-CCSD(T), что указывает на улучшенную масштабируемость и потенциальную эффективность для систем большего размера.
Влияние на системы без запрещенной зоны и перспективы развития
Точное моделирование электронной корреляции имеет первостепенное значение при изучении систем с отсутствием запрещенной зоны (Gapless Systems). В этих материалах, таких как некоторые полуметаллы и сильно коррелированные электронные системы, даже незначительные взаимодействия между электронами оказывают определяющее влияние на их физические свойства. В отличие от традиционных изоляторов или полупроводников, где энергия запрещенной зоны эффективно разделяет электронные состояния, в Gapless Systems электронные возбуждения могут происходить при сколь угодно малых энергиях, делая их чрезвычайно чувствительными к деталям электронных взаимодействий. Поэтому, для адекватного описания и предсказания свойств этих материалов, необходимо использовать методы, способные точно учитывать эффекты электронной корреляции, что представляет собой серьезную вычислительную задачу.
В рамках теории сцепленных кластеров, приближение замороженных ядер часто используется для упрощения вычислений, однако в системах с отсутствием запрещенной зоны, известных как gapless systems, это может приводить к существенным погрешностям. Данное приближение, исключая электроны внутренней оболочки из корреляционных расчетов, может недооценивать важные взаимодействия, влияющие на свойства материала. В подобных системах, где даже слабые электронные корреляции играют решающую роль, игнорирование вклада электронов внутренней оболочки может привести к неточным результатам, снижая надежность предсказаний свойств и поведения материала. Поэтому, для получения достоверных данных о gapless системах, необходимо тщательно оценивать влияние приближения замороженных ядер и, при необходимости, использовать более точные, хотя и вычислительно затратные, методы.
Современные вычислительные методы, такие как Стохастическая Тензорная Контракция (STC), открывают новые перспективы для моделирования сложных материалов и молекул. Исследования показывают, что STC-CCSD(T) демонстрирует более высокую точность по сравнению с DLPNO-CCSD(T) в ряде тестовых задач, что указывает на его потенциальное превосходство в расчетах, требующих высокой степени точности. Этот прогресс может привести к существенному улучшению в понимании свойств материалов, особенно в системах с узкой запрещенной зоной, где даже незначительные взаимодействия играют ключевую роль. В перспективе, STC-методы способны превзойти традиционную Теорию Функционала Плотности (DFT) в решении определенных задач, предлагая более надежный и точный инструмент для материаловедения и химии.
![Сравнение методов STC-CCSD(T), DLPNO-CCSD(T) и точного CCSD(T) на наборе из 20 реалистичных молекул, включая октаметилциклобутан (OMCB), [2.2]парациклофан (PCP) и дезоксицитидинмонофосфат (DCMP), показывает, что DLPNO-CCSD(T) с настройками NormalPNO и TightPNO обеспечивает сравнимую точность с точным CCSD(T) при меньших вычислительных затратах, а целевая погрешность STC-CCSD(T) установлена на уровне 0.2 ккал/моль.](https://arxiv.org/html/2602.17158v1/x7.png)
Исследование демонстрирует, что традиционные методы квантовой химии сталкиваются с экспоненциальным ростом вычислительных затрат при увеличении размера молекулярной системы. Предложенный подход — стохастическое сжатие тензоров — пытается обойти эту проблему, жертвуя некоторой точностью ради значительного снижения вычислительной сложности. Это напоминает о высказывании Стивена Хокинга: «Чем больше мы узнаем, тем больше понимаем, как мало мы знаем». Действительно, стремление к абсолютной точности в моделировании сложных систем часто оказывается непомерно дорогим, и необходимо искать компромиссы. Данная работа, фокусируясь на методе сжатия тензоров для теории сцепленных кластеров, иллюстрирует эту дилемму, предлагая прагматичный путь к достижению приемлемой точности при разумных вычислительных ресурсах. Ведь, как известно, упрощение — не всегда признак невежества, а иногда — признак мудрости.
Что дальше?
Представленный подход к стохастическому сжатию тензоров, безусловно, снижает вычислительную нагрузку, но не стоит обманываться кажущейся простотой. Каждая метрика — это идеология в disguise. Уменьшение сложности до масштаба теории среднего поля — это приятно, но вопрос в том, насколько глубоко можно углубиться в эту оптимизацию, не потеряв при этом точность. Если показатели растут, значит, кто-то неправильно измеряет. Необходимо тщательно изучить влияние стохастической природы метода на сходимость и стабильность расчетов, особенно для сложных молекулярных систем.
Очевидным направлением для дальнейших исследований является разработка более эффективных стратегий снижения дисперсии. Простое увеличение числа стохастических выборок — это не решение, а лишь временная отсрочка проблемы. Поиск алгоритмов, которые учитывают локальность тензорных связей, представляется особенно перспективным. В конце концов, сама природа квантовой механики предполагает, что не все степени свободы одинаково важны.
И, пожалуй, самое главное — необходимо отбросить иллюзию универсальности. Метод, успешно работающий для одной молекулы или одного класса молекул, может оказаться совершенно неэффективным для другой. Квантовая химия — это не сборник рецептов, а скорее искусство компромисса между точностью, скоростью и здравым смыслом. Данные не лгут — но люди, их интерпретирующие, часто фантазируют.
Оригинал статьи: https://arxiv.org/pdf/2602.17158.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Временная запутанность: от хаоса к порядку
- Улучшение точности квантовых сенсоров: новый подход к подавлению шумов
- Квантовое программирование: Карта развивающегося мира
- Предел возможностей: где большие языковые модели теряют разум?
- ЭКГ-анализ будущего: От данных к цифровым биомаркерам
- Резонансы в тандеме: Управление светом в микрорезонаторах
- Сердце музыки: открытые модели для создания композиций
- Квантовый шум: за пределами стандартных моделей
- Квантовые кольца: новые горизонты спиновых токов
- Искусственный разум и квантовые данные: новый подход к синтезу табличных данных
2026-02-20 17:14