Автор: Денис Аветисян
Исследователи демонстрируют, что использование симметрии в архитектуре нейронных сетей позволяет значительно улучшить точность и эффективность моделей, изучающих квантово-механические свойства молекул.
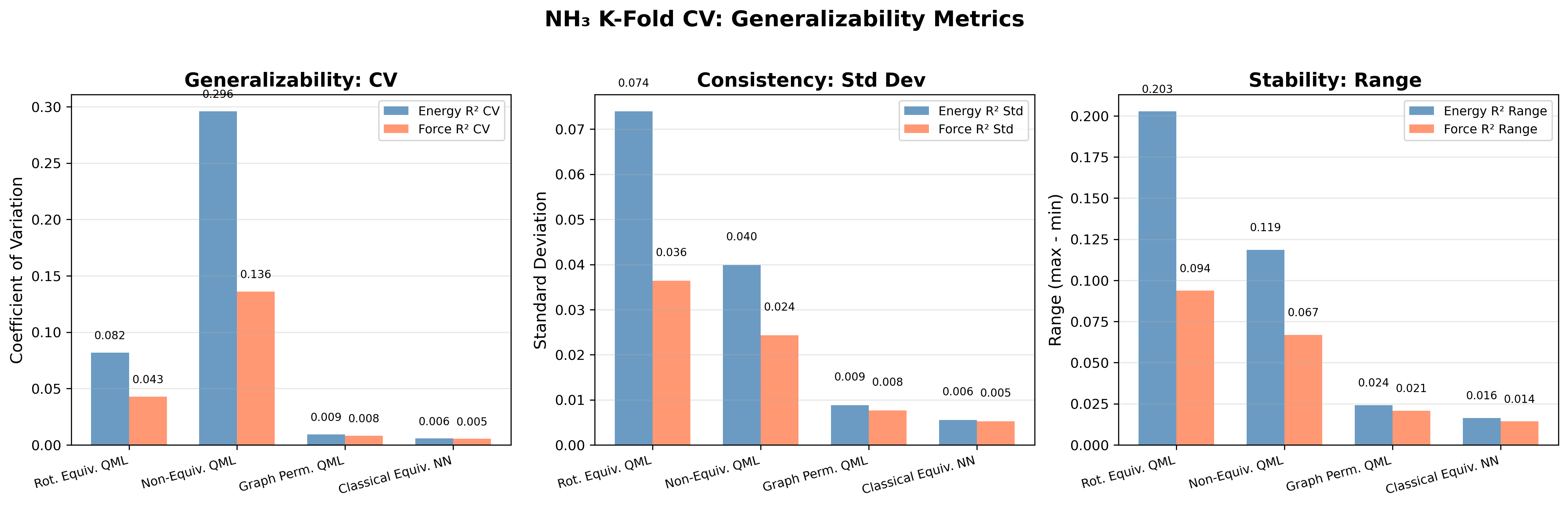
В статье показано, что учет пермутационной и геометрической симметрии в квантовом машинном обучении приближает производительность к классическим моделям, требуя при этом значительно меньше параметров.
Несмотря на растущий интерес к квантовому машинному обучению, сохраняется проблема обобщающей способности моделей при работе с геометрическими данными, характерными для молекулярной физики. В работе ‘PERM EQ x GRAPH EQ: Equivariant Neural Networks for Quantum Molecular Learning’ сравнивается эффективность различных моделей геометрического квантового машинного обучения, включая варианты с симметрией, а также встраивание графовых представлений. Показано, что использование пермутационной симметрии в графовых квантовых нейронных сетях значительно улучшает обобщающую способность моделей, приближая их к производительности классических аналогов при меньшем количестве параметров. Какие перспективы открываются для дальнейшего развития квантовых моделей, учитывающих симметрии молекулярных систем и позволяющих эффективно решать задачи молекулярного моделирования?
Симметрия и Пределы Традиционного Квантового Моделирования
Точность квантовых симуляций молекулярных систем сопряжена со значительными вычислительными затратами, которые зачастую растут экспоненциально с увеличением размера моделируемой системы. Это связано с тем, что для описания квантового состояния даже относительно небольших молекул требуется экспоненциальное количество параметров, что делает прямые вычисления практически невозможными для систем, содержащих более нескольких десятков атомов. Например, для точного описания $N$ не взаимодействующих спинов требуется $2^N$ комплексных чисел. По мере усложнения молекулярной структуры и появления электронных корреляций, эта экспоненциальная зависимость становится еще более выраженной, что обуславливает необходимость разработки новых алгоритмов и методов, направленных на снижение вычислительной сложности и повышение масштабируемости квантовых симуляций.
Традиционные методы квантово-химического моделирования, такие как метод Хартри-Фока и метод связанных кластеров с одинарными и двойными возбуждениями (CCSD), демонстрируют высокую точность при описании многих молекулярных систем. Однако, их применимость ограничена в случаях, когда электроны в молекуле сильно коррелированы, то есть их взаимодействие нельзя описать в рамках упрощенных приближений. В подобных ситуациях, возникающих в системах с раскрытыми оболочками или при наличии сильных электронных взаимодействий, требуются значительно более сложные вычисления. Кроме того, моделирование молекул со сложной геометрией, например, содержащих переходные металлы или большие кластеры, также представляет серьезную вычислительную проблему для этих методов, поскольку количество необходимых параметров и интегралов быстро растет с увеличением размера системы и числа атомов. Это приводит к экспоненциальному увеличению вычислительных затрат и ограничивает возможность исследования более сложных и реалистичных молекулярных систем.
Исследования показывают, что использование присущих молекулярным системам симметрий предоставляет эффективный способ снизить вычислительную сложность и повысить масштабируемость квантовых симуляций. Вместо обработки всех возможных конфигураций системы, алгоритмы могут быть разработаны для учета только тех, которые не нарушают симметрии молекулы. Это значительно сокращает размер пространства состояний, с которым необходимо работать, и позволяет моделировать более крупные и сложные системы при тех же вычислительных ресурсах. Например, в молекулах с высокой степенью симметрии, таких как $C_{nv}$ или $D_{nh}$ группы, количество независимых переменных, необходимых для описания волновой функции, резко уменьшается. Применение теории групп и соответствующих алгоритмов позволяет эффективно использовать эти симметрии, что приводит к экспоненциальному снижению вычислительных затрат и открывает новые возможности для моделирования сложных химических процессов и материалов.

Геометрическое Квантовое Машинное Обучение: Использование Симметрии для Эффективности
Геометрическое квантовое машинное обучение (ГКМО) представляет собой подход к построению квантовых моделей, который учитывает симметрии, присущие данным. Вместо обработки данных как абстрактных векторов, ГКМО использует принципы геометрии и теории групп для представления данных в виде симметричных тензоров. Это позволяет создавать квантовые схемы, инвариантные относительно определенных преобразований, таких как вращения или перестановки. Такой подход обеспечивает более эффективное представление данных, поскольку позволяет избежать избыточности и сконцентрироваться на существенных признаках, сохраняющихся при симметричных преобразованиях. Использование симметрий также упрощает процесс обучения модели, снижая требования к вычислительным ресурсам и объему обучающей выборки.
Геометрическое квантовое машинное обучение (ГКМО) использует математический аппарат теории групп для построения симметрийно-адаптированных представлений молекулярных данных. В частности, ГКМО оперирует с группами Ли, такими как $SO(3)$ (группа вращений в трехмерном пространстве) и $E(3)$ (группа Евклида, включающая вращения и трансляции), а также их алгебрами Ли, например, $SU(2)$ и $SO(3)$. Применение этих концепций позволяет декомпозировать молекулярные данные на неприводимые представления групп симметрии, что обеспечивает эффективное кодирование информации, сохраняющее инвариантность относительно симметричных преобразований и снижает вычислительную сложность модели.
В геометрическом квантовом машинном обучении (GQML) использование ограничений симметрии позволяет существенно сократить количество параметров, необходимых для представления сложного квантового состояния. Вместо обучения всем возможным параметрам, модель обучается лишь тем, которые соответствуют симметриям данных, что приводит к снижению вычислительной сложности и уменьшению потребности в обучающих данных. Например, для молекулярных данных, использование симметрий вращения и отражения позволяет сократить размер пространства параметров, необходимого для описания волновой функции, что особенно важно для работы с большими молекулами и сложными химическими системами. Это снижение размерности не только ускоряет процесс обучения, но и повышает обобщающую способность модели, уменьшая риск переобучения и улучшая ее производительность на новых, ранее не встречавшихся данных.

Симметрия-Эквивариантные Квантовые Нейронные Сети: Практическая Реализация
Симметрически-эквивариантные квантовые нейронные сети (SEQNN) представляют собой конкретную реализацию принципов графического квантового машинного обучения (GQML), разработанную для работы с молекулярными данными, представленными в виде графов. Эти сети используют структуру графа для кодирования информации о соединениях атомов и их свойствах, что позволяет эффективно обрабатывать сложные молекулярные структуры. В отличие от традиционных нейронных сетей, SEQNN разработаны для сохранения предсказаний при изменениях порядка атомов в графе или ориентации молекулы в пространстве, что критически важно для задач молекулярного моделирования и открытия новых материалов. Данный подход позволяет существенно сократить количество параметров модели и повысить её обобщающую способность при работе с различными молекулярными структурами.
Симметрия перестановочной инвариантности и вращательной эквивариантности являются ключевыми принципами построения симметричных квантовых нейронных сетей. Перестановочная инвариантность гарантирует, что предсказания сети не изменяются при перестановке атомов в молекуле, что критически важно для обработки молекулярных графов, где порядок атомов произволен. Вращательная эквивариантность обеспечивает, что предсказания преобразуются корректно при вращении всей молекулы в пространстве; например, энергия молекулы должна вращаться вместе с ней. Реализация этих свойств достигается за счет использования специальных слоев и функций активации, учитывающих симметрии молекулярной структуры, что повышает точность и обобщающую способность модели при предсказании молекулярных свойств.
Квантовые графовые нейронные сети (КГНС) используют методы встраивания данных для преобразования молекулярных графов в квантовые состояния, пригодные для обработки в симметрически-эквивариантных квантовых нейронных сетях (СЭКНС). Процесс встраивания включает отображение атомов и связей графа в векторы признаков, которые затем кодируются в квантовые биты (кубиты). Конкретные методы встраивания варьируются, но обычно включают использование тензорных представлений для кодирования информации о связях и окружении атомов, что позволяет сохранить структурную информацию молекулы при переходе в квантовое представление. Полученное квантовое состояние описывается волновой функцией $ |\psi \rangle $, которая является входными данными для последующей обработки СЭКНС.
Валидация, Данные и Эпоха NISQ
Оценка способности модели SEQNN к обобщению, то есть к корректной работе с новыми, ранее не виденными данными, требует применения методов перекрестной проверки. Этот процесс позволяет надёжно установить, насколько хорошо модель предсказывает свойства молекул, не участвовавших в её обучении. Для количественной оценки используются различные метрики, такие как $R^2$ (коэффициент детерминации), средняя абсолютная ошибка (Mean Absolute Error) и среднеквадратичная ошибка (Root Mean Squared Error). Высокие значения $R^2$ и низкие значения ошибок указывают на то, что модель обладает высокой точностью и способна эффективно предсказывать свойства новых молекулярных структур, что особенно важно для применения в квантовой химии и материаловедении.
Для успешного обучения моделей квантового машинного обучения, в частности, SEQNN, критически важны высококачественные данные, полученные методами ab initio квантовой химии. Эти данные, генерируемые с использованием теории функционала плотности (DFT) и уравнений Kohn-Sham, предоставляют точные решения $Schrödinger$ уравнения для молекулярных систем. Точность и разнообразие таких данных напрямую влияют на способность модели к обобщению и предсказанию свойств новых молекул. В частности, использование DFT позволяет вычислить электронную структуру молекул и получить информацию о энергии, геометрии и других важных характеристиках, что необходимо для обучения моделей, способных эффективно моделировать химические процессы и открывать новые материалы.
Исследование показало, что включение пермутационной эквивариантности в модели квантового машинного обучения (QML) значительно повышает их способность к обобщению, особенно при работе с молекулами сложной геометрией. В отличие от традиционных QML-моделей, не учитывающих симметрию молекулярных структур, разработанный подход позволяет учитывать перестановки атомов, обеспечивая инвариантность предсказаний относительно этих перестановок. Это достигается за счет использования специальных слоев и функций активации, которые учитывают симметрию графа молекулы. Результаты демонстрируют, что производительность полученных QML-моделей сопоставима с производительностью классических эквивариантных моделей машинного обучения, что открывает новые возможности для точного и эффективного моделирования молекулярных свойств и реакций. Применение данного метода особенно актуально для изучения сложных органических молекул и материалов, где учет симметрии критически важен для получения достоверных результатов.

За Пределами NISQ: Будущее Симметрия-Уважающего Квантового Машинного Обучения
Исследования показывают, что применение усовершенствованных функций активации, таких как Sigmoid-Linear-Unit (SLU), и функций потерь, например, Huber Loss, в симметричных квантовых нейронных сетях (SEQNN) способно значительно повысить их производительность и устойчивость к шумам. В отличие от традиционных функций активации, SLU обеспечивает более эффективное распространение градиентов, что критически важно для обучения глубоких квантовых сетей. Huber Loss, в свою очередь, менее чувствительна к выбросам в данных, чем среднеквадратичная ошибка, что делает SEQNN более надежными в реальных условиях, где данные часто бывают зашумлены или неполны. Эти усовершенствования открывают возможности для создания более точных и эффективных квантовых моделей машинного обучения, способных решать сложные задачи в различных областях науки и техники, например, в области анализа данных и распознавания образов.
Перспективным направлением развития квантового машинного обучения является расширение области его применения за пределы традиционных задач. Особый интерес представляет использование методов, учитывающих симметрии, в таких областях, как материаловедение и разработка лекарственных препаратов. В материаловедении, алгоритмы группового квантового машинного обучения (GQML) способны эффективно моделировать сложные электронные структуры и предсказывать свойства новых материалов, учитывая их кристаллическую симметрию. В фармацевтике, GQML может использоваться для предсказания взаимодействия молекул лекарств с биологическими мишенями, что существенно ускорит процесс открытия новых лекарственных средств. Такой подход позволяет существенно сократить объем вычислительных ресурсов, необходимых для моделирования сложных систем, и повысить точность предсказаний, что делает его особенно актуальным для решения задач, требующих высокой степени детализации и учета множества факторов.
По мере развития кванзитного оборудования, симметрия-сохраняющее квантовое машинное обучение станет ключевым инструментом для реализации полного потенциала квантового моделирования и открытий. Особенность подхода заключается в эффективном использовании симметрий физических систем, что позволяет существенно снизить вычислительные затраты и повысить точность моделирования сложных процессов. Вместо обработки огромного объема данных без учета внутренних закономерностей, алгоритмы, учитывающие симметрии, способны концентрироваться на наиболее важных параметрах, значительно ускоряя обучение и повышая устойчивость к шуму. Это особенно важно при моделировании молекул, материалов и других квантовых систем, где симметрии играют фундаментальную роль. Ожидается, что в будущем симметрия-сохраняющее квантовое машинное обучение станет незаменимым инструментом для ученых, работающих в области химии, физики материалов и разработки новых лекарственных препаратов, открывая возможности для решения задач, недоступных классическим вычислительным методам.
Исследование демонстрирует, что включение пермутационной эквивариантности графов в квантовое машинное обучение позволяет создавать модели, обладающие большей обобщающей способностью при изучении молекулярной физики. Это, по сути, признание того, что структура системы, её внутренние связи, определяют её поведение. Как заметил Джон фон Нейман: «В науке не бывает абсолютной точности, только степени неопределённости». Данная работа подтверждает эту мысль, показывая, что даже в квантовых моделях, учет симметрий и связей между компонентами системы (в данном случае, атомами в молекуле) критически важен для получения надежных результатов и приближения к производительности классических методов при значительно меньшем количестве параметров. Системы не просто существуют, они развиваются в соответствии со своей внутренней логикой, и игнорирование этой логики приводит к неизбежным ошибкам.
Что Дальше?
Представленная работа демонстрирует не столько построение модели, сколько культивирование способности к обобщению в пространстве молекулярных представлений. Упор на пермутационную эквивариантность — это признание того, что хаос не является сбоем, а языком природы, и попытка говорить на нём. Однако, достижение производительности, сопоставимой с классическими методами, при меньшем количестве параметров — это лишь первый шаг. Гарантий стабильности здесь нет, лишь договор с вероятностью.
Следующим этапом представляется не столько расширение архитектуры, сколько углубление понимания лежащих в основе принципов геометрического машинного обучения. Необходимо исследовать, как симметрии, выходящие за рамки пермутаций, могут быть эффективно включены в модели квантового машинного обучения. Стабильность — это иллюзия, которая хорошо кэшируется, но истинное развитие требует преодоления её ложного комфорта.
В конечном счете, задача состоит не в создании идеальной модели для конкретной задачи, а в разработке экосистемы, способной адаптироваться и эволюционировать вместе с данными. Каждый архитектурный выбор — это пророчество о будущем сбое, и признание этого — ключ к созданию действительно устойчивых и гибких систем.
Оригинал статьи: https://arxiv.org/pdf/2512.05475.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Карта ошибок: Анатомия сбоев больших языковых моделей
- Ядерный синтез и Искусственный Интеллект: Новый подход к проектированию реакторов
- Надежность ускорителей: от замысла до реализации
- Квантовые нейросети для реалистичной 3D-визуализации
- Nemotron Nano V2 VL: Зрение и язык в новом формате
- Знания в графах: как улучшить ответы больших языковых моделей
- Упорядоченный разум: Как языковые модели учатся справляться с длинными текстами
- Искусственный интеллект, действующий по цели: эволюция архитектуры
- Накапливая опыт: мультимодальные агенты, которые учатся на ходу
- Искуственный интеллект: хрупкость смысла в сложных задачах
2025-12-08 18:56