Автор: Денис Аветисян
Исследование представляет комплексную методологию и набор эталонных систем для оценки и повышения эффективности квантовых алгоритмов в решении задач сильной электронной корреляции.
Предложенная методика ADAPT-GCIM и набор химических систем позволяют оценить прогресс в достижении квантового превосходства для моделирования сложных молекул, включая соединения актинидов.
Несмотря на значительный прогресс в квантовых вычислениях, демонстрация практической пользы в решении реальных химических задач остается сложной задачей. В работе ‘Chemically decisive benchmarks on the path to quantum utility’ представлен тщательно подобранный набор химических систем, предназначенный для оценки и улучшения работы квантовых алгоритмов в расчетах сильно коррелированных электронных структур. Показано, что разработанный рабочий процесс, основанный на алгоритме ADAPT-GCIM, обеспечивает высокую точность в различных корреляционных режимах, выявляя при этом общие ограничения существующих подходов. Смогут ли эти эталонные тесты ускорить разработку квантовых алгоритмов, способных эффективно решать сложные химические проблемы и приблизить эру квантовой химии?
Электронная корреляция: вызов современной квантовой химии
Точное описание взаимодействия между электронами — электронная корреляция — представляет собой фундаментальную задачу в квантовой химии. Взаимодействие электронов выходит далеко за рамки простого кулоновского отталкивания; оно включает в себя сложные динамические эффекты, возникающие из-за того, что каждый электрон испытывает мгновенное влияние со стороны всех остальных. Игнорирование или неточное описание этих корреляционных эффектов приводит к существенным погрешностям при расчете энергии и свойств молекул, особенно в случаях, когда электроны сильно взаимодействуют, например, в переходных металлах или при разрыве и образовании химических связей. Понимание и адекватное моделирование электронной корреляции критически важно для предсказания химического поведения веществ и разработки новых материалов с заданными свойствами, что делает эту область активным направлением исследований в современной квантовой химии и вычислительной физике.
Традиционные методы квантово-химических расчетов, такие как Полная Конфигурационная Взаимодействие (CASCI), сталкиваются с серьезными ограничениями при моделировании даже умеренно сложных молекул. Проблема заключается в экспоненциальном росте вычислительных затрат с увеличением числа электронов и атомных орбиталей, учитываемых в расчете. Это означает, что для каждого добавленного электрона или орбитали, время и ресурсы, необходимые для получения точного результата, растут не линейно, а экспоненциально, что делает применение CASCI к большим системам практически невозможным. Таким образом, несмотря на высокую точность, CASCI становится непрактичным инструментом для исследования сложных химических процессов и материалов, что стимулирует поиск более эффективных и масштабируемых методов учета электронной корреляции.
Ограничения в точном моделировании электронных взаимодействий существенно препятствуют исследованию сложных химических систем. Невозможность адекватно описать поведение электронов в молекулах среднего и большого размера приводит к неточностям в предсказании их свойств и реакционной способности. Это особенно критично для таких областей, как разработка новых материалов, катализ и понимание биологических процессов, где даже незначительные погрешности в расчетах могут привести к ошибочным выводам и неэффективным экспериментам. Точность моделирования напрямую влияет на возможность предсказать стабильность молекул, энергии реакций и спектральные характеристики, что делает решение проблемы электронной корреляции ключевым для прогресса в современной химии и физике.
ADAPT-GCIM: гармонизируя точность и вычислительную эффективность
Алгоритм ADAPT-GCIM представляет собой перспективный подход к квантово-химическим расчетам, объединяющий методы генераторных координат (Generator Coordinate Methods, GCM) и вариационные квантовые решатели (Variational Quantum Eigensolvers, VQE). GCM позволяет эффективно аппроксимировать волновые функции сложных систем путем разложения на линейные комбинации функций, полученных при различных деформациях геометрии молекулы. VQE, в свою очередь, использует квантовые вычисления для оптимизации параметров вариационного ответа, минимизируя энергию системы. Комбинация этих методов позволяет эффективно решать задачу нахождения основного состояния молекулы, используя возможности как классических, так и квантовых вычислительных ресурсов. Такой гибридный подход направлен на снижение вычислительной сложности, сохраняя при этом точность расчетов.
Ключевым элементом алгоритма ADAPT-GCIM является интеллектуальный выбор «активной области» — ограниченного набора орбиталей — посредством инструмента ActiveSpaceFinder. Этот подход позволяет целенаправленно сконцентрировать вычислительные ресурсы на наиболее важных областях электронной структуры, минимизируя тем самым общие вычислительные затраты. ActiveSpaceFinder анализирует вклад различных орбиталей в корреляционные эффекты и выбирает только те, которые оказывают существенное влияние на энергетические уровни и свойства молекулы. Такая адаптивная стратегия позволяет проводить расчеты для более крупных систем, сохраняя при этом необходимую точность и избегая избыточных вычислений для незначимых орбиталей.
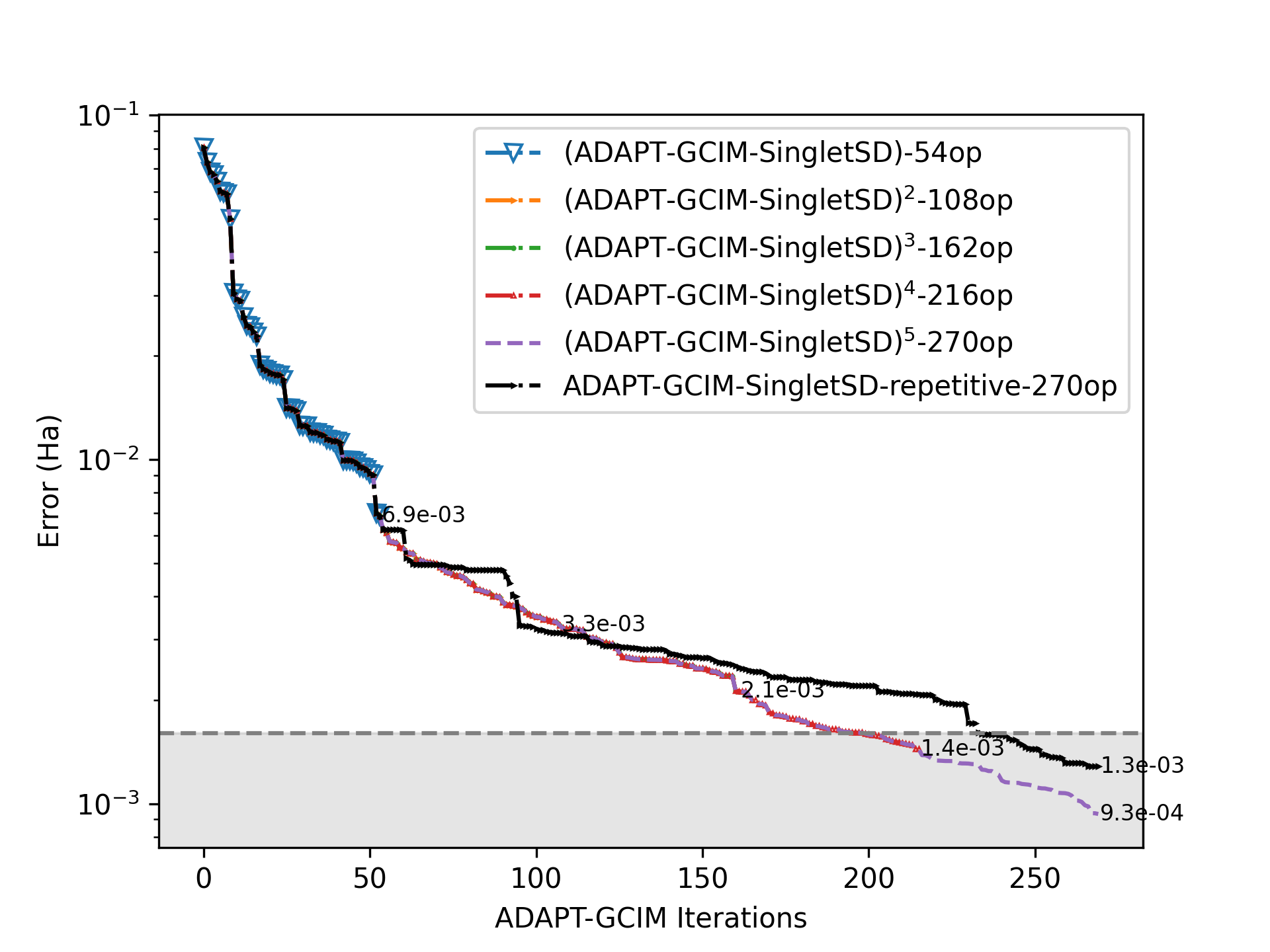
Адаптивная стратегия, реализованная в ADAPT-GCIM, позволяет существенно снизить вычислительные затраты при сохранении ключевых эффектов электронной корреляции. Это достигается за счет динамической оптимизации размера и состава ‘активной области’ — набора орбиталей, используемых в расчетах. Сокращение активной области уменьшает экспоненциальную сложность вычислений, позволяя проводить расчеты для систем большего размера, чем это возможно при использовании традиционных методов. При этом, благодаря интеллектуальному подбору активной области, обеспечивается достижение так называемой ‘химической точности’ — ошибок не превышающих 1.59 \times 10^{-3} \text{ Ha}, что критически важно для надежного моделирования химических свойств и реакций.

Конструирование эффективных квантовых анзацев для точных расчетов
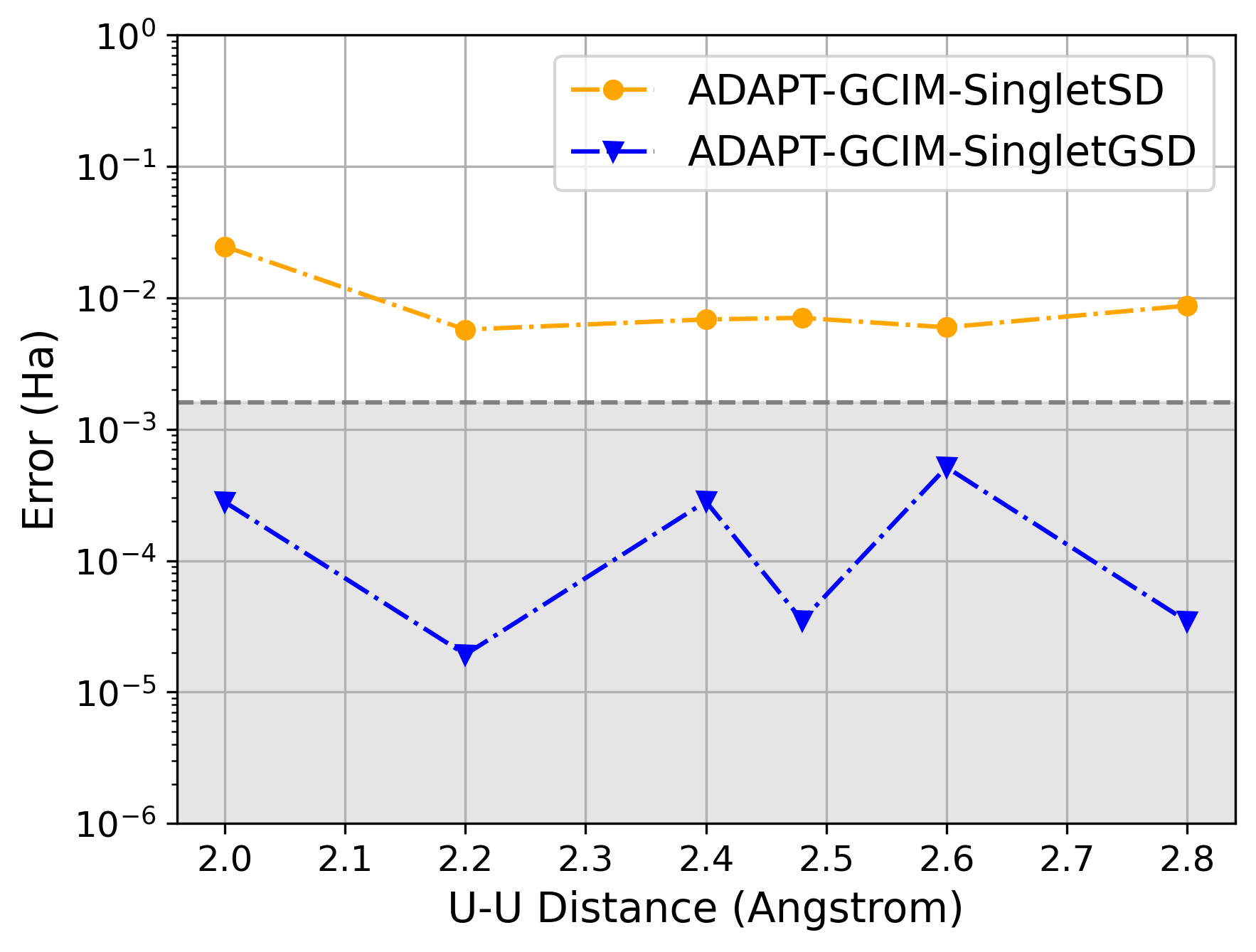
Метод ADAPT-GCIM использует базисы возбуждений, такие как singletSD и singletGSD, для построения квантового ‘анзаца’ — параметризованной волновой функции, применяемой в вариационном расчете. Базис singletSD состоит из одинарных и двойных возбуждений, в то время как singletGSD включает в себя одинарные, двойные и тройные возбуждения, что позволяет более гибко описывать электронную корреляцию в выбранном активном пространстве. Выбор конкретного базиса возбуждений влияет на выразительность и вычислительную стоимость анзаца, определяя его способность точно моделировать электронную структуру исследуемой системы. Конструкция анзаца на основе этих базисов является ключевым этапом в алгоритме ADAPT-GCIM, обеспечивающим эффективное приближение к решению уравнения Шредингера для молекулярных систем.
Основы возбуждений, такие как singletSD и singletGSD, предоставляют гибкую структуру для описания ключевых эффектов электронной корреляции в пределах выбранного активного пространства. Эти основы позволяют строить волновые функции, учитывающие взаимодействия между электронами, выходящие за рамки независимых частиц. В частности, singletSD описывает одинарные и двойные возбуждения электронов, а singletGSD включает в себя дополнительные возбуждения, что обеспечивает более точное моделирование электронной структуры и, как следствие, более точные предсказания свойств молекул и материалов. Выбор подходящей основы возбуждений критически важен для эффективного использования вариационного принципа в квантовых вычислениях.
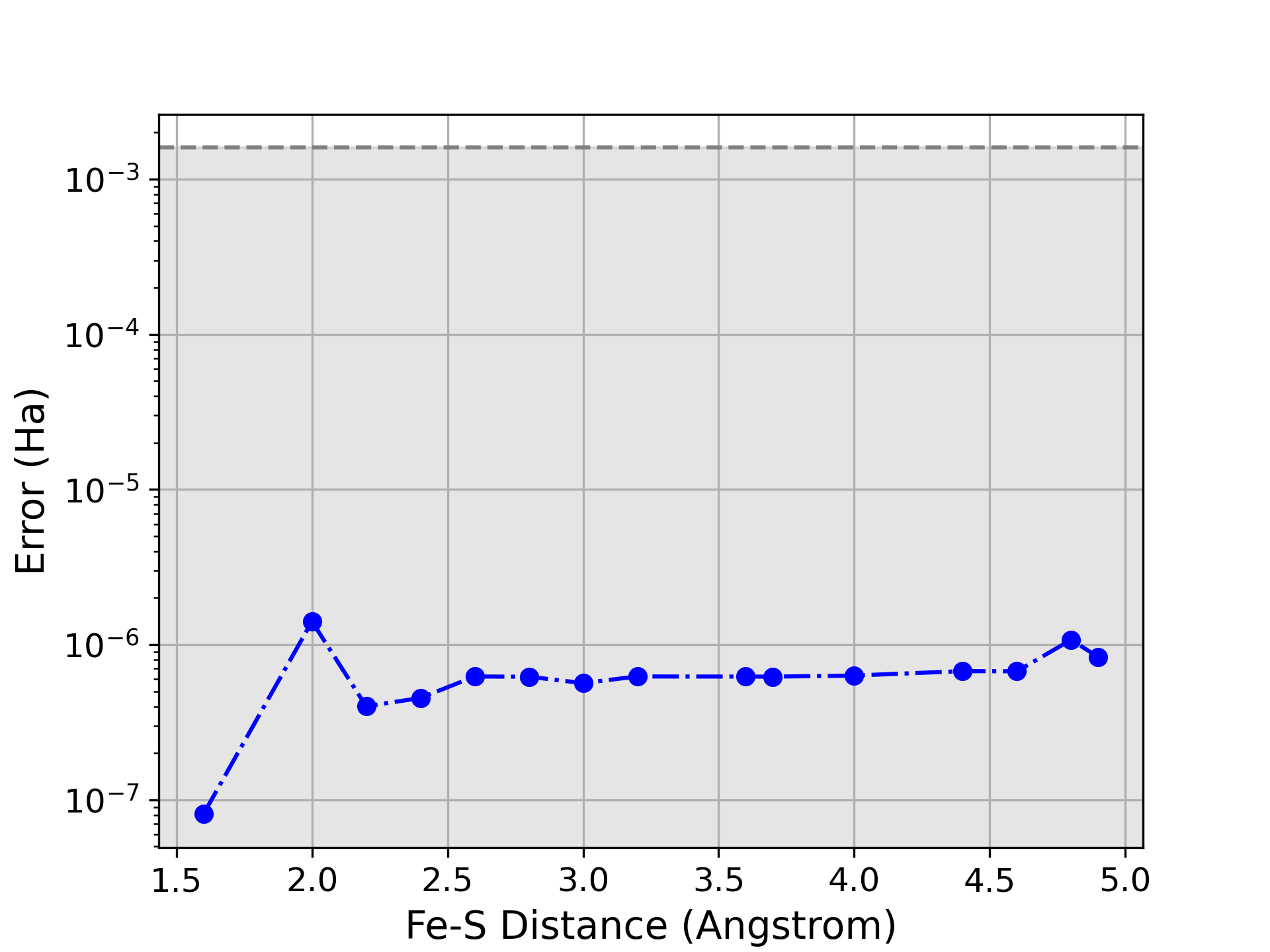
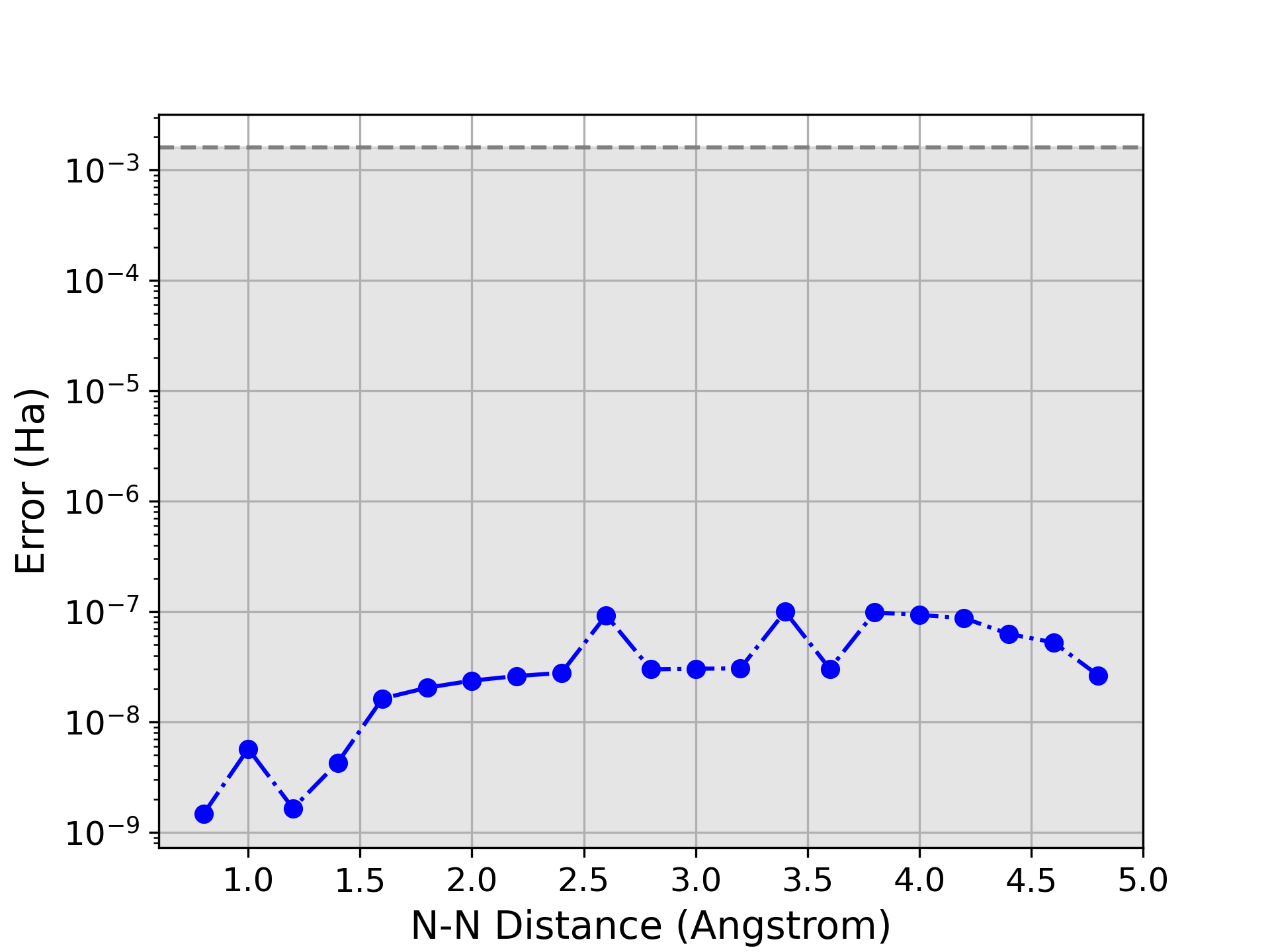
Валидация метода ADAPT-GCIM была проведена посредством эталонных расчетов для диатомных молекул, таких как N2, FeS и U2, а также для кластера [2Fe-2S]. Результаты показали, что метод способен точно предсказывать свойства исследуемых систем, стабильно достигая химической точности, определяемой как погрешность менее 1.59 \times 10^{-3} \text{ Ha}. Данные тесты подтверждают надежность и применимость ADAPT-GCIM для квантово-химических расчетов, требующих высокой точности.
Квантовое преимущество в моделировании молекул: перспективы и достижения
Применение алгоритма ADAPT-GCIM к системам возрастающей сложности, в частности к FeS-кластеру, позволяет оценить его масштабируемость и производительность. Исследования демонстрируют, что данный подход способен эффективно обрабатывать системы, требующие точного моделирования сильных корреляционных эффектов, что особенно важно для понимания поведения биоорганических систем. Оценка производительности на FeS-кластере, содержащем значительное количество электронов и орбиталей, указывает на перспективность метода для решения задач, недоступных классическим вычислительным подходам. Результаты этих исследований служат важным шагом на пути к реализации квантового превосходства в молекулярном моделировании и открывают возможности для разработки новых материалов и лекарственных препаратов, используя активное пространство (6e, 6o).
Способность алгоритма точно моделировать эффекты сильной корреляции имеет первостепенное значение для понимания поведения бионеорганических систем. В таких системах, как железо-серные кластеры, взаимодействие между электронами становится доминирующим, и традиционные методы квантовой химии часто дают неточные результаты. Алгоритм, учитывая эти сложные электронные корреляции, позволяет достоверно предсказывать энергетические уровни, магнитные свойства и реакционную способность, что необходимо для понимания роли этих кластеров в биологических процессах, например, в переносе электронов и катализе. H = \sum_{i} h_{i} + \sum_{i<j} g_{ij}[="" latex]="" p="" возможности="" действия="" для="" и="" катализаторов="" квантовом="" механизмов="" на="" новых="" открывает="" подход="" понимания="" разработки="" такой="" уровне.<="" ферментов=""></p> <p>Успешное применение алгоритма к сложным системам, таким как железосерные кластеры, открывает перспективы для решения более амбициозных задач в материаловедении и разработке лекарственных препаратов. С использованием активного пространства [latex](6e, 6o), квантовые вычисления способны моделировать сильные корреляционные эффекты, критически важные для понимания поведения этих биоорганических систем и предсказания свойств новых материалов. Этот подход обещает революционизировать химические инновации, позволяя проводить точные и эффективные расчеты, недоступные классическим методам, и ускорить открытие новых соединений с заданными характеристиками.
Исследование, представленное в данной работе, демонстрирует важность систематического подхода к анализу корреляционного ландшафта в квантовой химии. Авторы предлагают алгоритм ADAPT-GCIM, позволяющий эффективно отбирать активное пространство для вариационного квантового эйнсольвера, что критически важно для моделирования сильно коррелированных систем. Как однажды заметил Ричард Фейнман: «Если вы не можете объяснить что-то простыми словами, значит, вы сами этого не понимаете». Данный принцип находит отражение в стремлении исследователей упростить сложные квантовые расчеты, выявляя ключевые закономерности и оптимизируя алгоритмы для достижения квантового преимущества в химии. Подобный подход позволяет не просто получить результат, но и понять лежащие в его основе физические принципы.
Куда же дальше?
Представленный анализ, хотя и демонстрирует ощутимый прогресс в оценке возможностей квантовых алгоритмов для решения сложных химических задач, не является финальной точкой, а скорее, новым рубежом. Особый интерес представляет не столько достижение точности, сколько понимание природы ошибок. Каждое отклонение от ожидаемого результата - это возможность выявить скрытые зависимости в корреляционном ландшафте, а также ограничения применяемых приближений. Следующим шагом видится разработка более адаптивных стратегий выбора активного пространства, способных динамически реагировать на изменяющиеся требования точности и вычислительных ресурсов.
Очевидным ограничением остается масштабируемость. Алгоритм ADAPT-GCIM, несмотря на свою эффективность, требует значительных вычислительных затрат. Поиск компромисса между точностью и скоростью, возможно, потребует обращения к новым архитектурам квантовых компьютеров или разработке гибридных классическо-квантовых подходов, способных эффективно использовать ресурсы обоих типов вычислений. Необходимо признать, что "квантовое превосходство" в химии - это не одномоментное достижение, а скорее, постепенное расширение области решаемых задач.
В конечном счете, истинная ценность подобных исследований заключается не в решении конкретных химических задач, а в углублении понимания фундаментальных принципов квантовой механики и её применения к сложным системам. Поиск "квантовой полезности" - это не просто техническая задача, но и философский поиск границ познания и возможностей моделирования реальности.
Оригинал статьи: https://arxiv.org/pdf/2601.10813.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Устойчивое обучение языковых моделей: новый подход к контролю стратегии
- Сердце музыки: открытые модели для создания композиций
- Самообучающиеся системы: новый подход к созданию многоагентных взаимодействий
- Квантовый усилитель света на чипе: новый уровень эффективности
- Квантовые системы в полуклассическом режиме: новый подход к моделированию
- Взгляд под капот: Анализ кода, сгенерированного нейросетями
- 💸 Великобритания тратит 500 миллионов фунтов стерлингов на квантовые технологии – может быть, кот Шрёдингера только что разбогател?
- Искусственный интеллект и архитектура будущего: новый виток эволюции
- Видео будущего: генерация длинных роликов без обучения
- Квантовое управление: от теории к практике
2026-01-19 10:42