Автор: Денис Аветисян
Исследователи продемонстрировали, что не зависящие от ориентации нейронные сети могут достигать сопоставимой точности и скорости с передовыми моделями для расчета межмолекулярных сил.
Разработанный подход позволяет создавать высокоточные и эффективные межмолекулярные потенциалы для крупномасштабного моделирования атомных систем.
Традиционные модели межатомных потенциалов, основанные на машинном обучении, зачастую ограничиваются строгим соблюдением физических законов сохранения и симметрий. В работе ‘Pushing the limits of unconstrained machine-learned interatomic potentials’ исследуется возможность отказа от этих ограничений при масштабировании моделей до больших объемов данных и параметров. Полученные результаты демонстрируют, что неконстрейнированные модели, обученные на крупных наборах данных, могут превосходить по точности и скорости констрейнированные аналоги, особенно в задачах оптимизации геометрии и динамики решеток. Возможно ли дальнейшее повышение эффективности и точности моделей межатомных потенциалов за счет разработки новых подходов к обучению и архитектуре сетей?
Основы вычислительной материаловедения: Межатомные взаимодействия
В основе вычислительной материаловедения лежит точное описание энергетического ландшафта атомных систем посредством межatomных потенциалов. Эти потенциалы, по сути, являются математическими функциями, которые позволяют предсказывать энергию системы атомов в зависимости от их взаимного расположения. От точности этих функций напрямую зависит достоверность моделирования различных материалов и предсказание их свойств. Разработка адекватных межatomных потенциалов — сложная задача, требующая учета квантово-механических эффектов, но с использованием принципов упрощения, таких как принцип локальности, становится возможным создание эффективных моделей, пригодных для масштабного моделирования структуры и поведения материалов. E = f(r_1, r_2, ..., r_N) — общая форма представления энергии системы N атомов в зависимости от их координат.
Принцип локальности, являясь краеугольным камнем при построении межатомных потенциалов, предполагает, что взаимодействие между атомами существенно убывает с расстоянием. Это упрощает задачу, позволяя рассматривать взаимодействие лишь с ближайшими соседями, вместо учета всех атомов в системе. Однако, такое упрощение требует тщательной разработки функциональной формы потенциала и точного определения параметров, чтобы адекватно описать физические свойства материала. Несмотря на кажущуюся простоту, корректное описание короткодействующих сил, включая ван-дер-ваальсовы взаимодействия и эффекты экранирования, критически важно для получения достоверных результатов в вычислительных материаловедческих исследованиях. Игнорирование этих нюансов может привести к значительным ошибкам в предсказании свойств материалов, таких как энергия связи, упругие характеристики и стабильность кристаллической структуры.
Для создания физически достоверных межатомных потенциалов, описывающих взаимодействие атомов в материалах, необходимо строгое соблюдение фундаментальных симметрий, в частности, инвариантности относительно перестановки атомов. Данное требование означает, что энергия системы не должна изменяться при любой перестановке идентичных атомов в её составе. Игнорирование этой симметрии приводит к некорректным результатам моделирования, искажая предсказываемые свойства материалов и вводя искусственные зависимости от порядка следования атомов в расчётах. Таким образом, инвариантность относительно перестановки является краеугольным камнем при разработке точных и надёжных межатомных потенциалов, обеспечивая соответствие результатов моделирования физической реальности и позволяя получать воспроизводимые и значимые данные.
![Предложенная архитектура рассчитывает общую энергию системы, суммируя атомные энергии <span class="katex-eq" data-katex-display="false">E_i</span> для каждого атома и его соседей <span class="katex-eq" data-katex-display="false">a_{i}</span> и <span class="katex-eq" data-katex-display="false">a_{ij}</span>, при этом веса внимания масштабируются для обеспечения плавности, как описано в работе [9].](https://arxiv.org/html/2601.16195v1/x1.png)
Моделирование поведения материалов: Основные вычислительные методы
Молекулярная динамика (МД) представляет собой численный метод, позволяющий моделировать движение атомов и молекул во времени. В основе метода лежит решение уравнений движения Ньютона для каждого атома в системе, где силы, действующие на атом, рассчитываются на основе межатомных потенциалов. Эти потенциалы описывают энергию взаимодействия между атомами в зависимости от их взаимного расположения. Решение этих уравнений, как правило, выполняется численно с использованием алгоритмов интегрирования, таких как Verlet или Leap-frog. МД позволяет изучать динамические процессы, такие как диффузия, фазовые переходы, механические свойства материалов и реакции, путем наблюдения за траекториями движения атомов во времени. Точность моделирования напрямую зависит от выбора межатомного потенциала и параметров моделирования, включая шаг интегрирования и температуру системы. F = ma — основное уравнение, используемое в МД.
Оптимизация геометрии — это вычислительный метод, использующий межatomные потенциалы для определения конфигурации структуры с минимальной энергией. В процессе оптимизации, алгоритмы итеративно изменяют положения атомов в модели, вычисляя энергию системы на каждом шаге. Цель состоит в том, чтобы найти такое расположение атомов, при котором энергия системы достигает минимума, что соответствует стабильной конфигурации. Этот процесс критически важен для предсказания равновесных структур материалов, анализа их устойчивости и определения свойств, зависящих от атомного расположения. Алгоритмы оптимизации геометрии часто используют градиентные методы или другие численные подходы для эффективного поиска минимума энергии на многомерной поверхности потенциальной энергии.
Метод Монте-Карло представляет собой вычислительный подход, использующий случайную выборку для исследования пространства конфигураций системы и расчета ее термодинамических свойств. В основе метода лежит генерация случайных перемещений атомов или молекул в моделируемой системе, при этом принятие или отклонение каждого перемещения определяется на основе энергетического изменения, вычисляемого с использованием межатомных потенциалов. Вероятность принятия перемещения обычно определяется функцией Больцмана exp(-ΔE/kT), где ΔE — изменение энергии, k — постоянная Больцмана, а T — температура. Повторяя этот процесс многократно, метод Монте-Карло позволяет эффективно исследовать пространство конфигураций и получать статистически значимые оценки термодинамических величин, таких как средняя энергия, теплоемкость и свободная энергия.
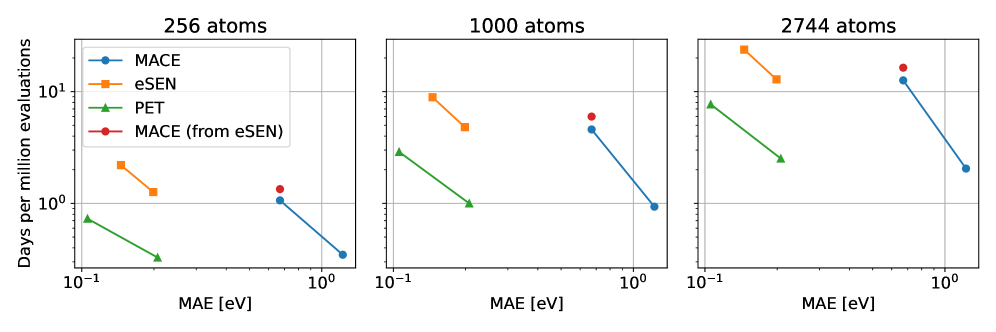
За пределами дифференцирования: Прямое предсказание сил
Традиционные методы вычисления сил, возникающих между атомами, основаны на дифференцировании потенциальной энергии. Этот процесс, требующий вычисления производных потенциальной функции по координатам атомов, может быть вычислительно затратным, особенно для сложных систем и больших моделей. Более того, численное дифференцирование подвержено ошибкам округления и чувствительно к выбору шага дифференцирования, что может приводить к неточностям в определении межмолекулярных сил. Погрешности, возникающие при дифференцировании, могут существенно влиять на результаты моделирования динамики молекулярных систем и предсказание их свойств.
Прямое предсказание сил (Direct Force Prediction) представляет собой альтернативный подход к вычислению межatomных сил, который обходит необходимость в дифференцировании потенциальной энергии. Вместо вычисления градиента потенциальной энергии для определения сил, данный метод непосредственно предсказывает значения сил, действующих между атомами. Это позволяет избежать вычислительных затрат, связанных с дифференцированием, а также снижает вероятность возникновения численных ошибок, что потенциально повышает эффективность моделирования атомных систем. Такой подход особенно актуален для сложных систем и больших масштабов моделирования, где традиционные методы, основанные на дифференцировании, могут оказаться вычислительно дорогостоящими или неточными.
В отличие от традиционных методов, основанных на расчете сил из потенциальных энергий и зависящих от консервативных сил, прямой прогноз сил позволяет учитывать и неконсервативные силы, расширяя возможности моделирования. Данный подход обеспечивает сопоставимую точность с передовыми моделями, такими как DPA-3.1-3M, что подтверждается результатами тестирования на бенчмарке matbench-discovery. Это позволяет моделировать системы, в которых неконсервативные силы играют значимую роль, например, процессы с диссипацией энергии или внешними воздействиями, не ограничиваясь потенциальными энергетическими поверхностями.
Исследование колебательных мод: Расчеты фононов
Расчеты фононов позволяют определить частоты и моды колебаний атомов в материале, что является фундаментальным для понимания его тепловых и акустических свойств. Эти колебания, или фононы, описывают коллективные движения атомов и напрямую влияют на способность материала проводить тепло и распространять звук. Изучение этих мод позволяет предсказать, как материал будет реагировать на изменения температуры, механическое воздействие и другие внешние факторы. Например, материалы с высокой частотой фононов обычно обладают более высокой теплопроводностью, в то время как определенные моды колебаний могут определять оптические свойства материала. Таким образом, понимание фононного спектра является ключевым для разработки материалов с заданными характеристиками в различных областях, от электроники до энергетики.
Традиционные методы вычисления фононов, определяющих колебательные свойства материалов, часто опираются на метод конечных разностей для аппроксимации производных. Этот подход, хотя и широко распространен, сопряжен со значительными вычислительными затратами. Для точного определения производных требуется выполнение большого числа расчетов для различных небольших смещений атомов в кристаллической решетке. С увеличением размера системы и сложности ее структуры, количество необходимых вычислений экспоненциально возрастает, что делает анализ больших и сложных материалов крайне трудоемким и ресурсозатратным. Таким образом, эффективность и масштабируемость расчетов фононов напрямую зависят от поиска более эффективных методов аппроксимации производных или применения альтернативных подходов, способных снизить вычислительную сложность.
Интеграция прямого предсказания сил в расчеты фононов позволяет значительно ускорить процесс моделирования колебательных свойств материалов и потенциально повысить точность результатов. Данный подход, основанный на предсказании сил, демонстрирует конкурентоспособную точность при анализе набора данных SPICE, используемого для проверки методов расчета фононов. В частности, полученные результаты указывают на возможность увеличения скорости вычислений в 2-3 раза по сравнению с консервативными моделями, что открывает перспективы для изучения более широкого спектра материалов и предсказания их тепловых и акустических характеристик с большей эффективностью. Это особенно важно для разработки новых материалов с заданными свойствами, требующих детального понимания их колебательного поведения.
Исследование, представленное в данной работе, демонстрирует, что не ограничивая модель пространственными вращениями, можно достичь сопоставимой точности и скорости вычислений, что открывает новые горизонты для крупномасштабного моделирования атомных систем. Этот подход, использующий графовые нейронные сети, ставит под вопрос традиционные методы разработки межatomных потенциалов. Как однажды заметил Альберт Эйнштейн: «Самое главное — это не переставать задавать вопросы». В контексте данной работы, отказ от жёстких ограничений на вращения — это, по сути, вопрос о том, как лучше всего представить физическую реальность в вычислительной модели. Масштабирование без проверки ценностей — преступление против будущего, и данное исследование подчеркивает необходимость постоянного поиска более эффективных и точных методов моделирования материалов.
Что дальше?
Представленная работа, демонстрируя жизнеспособность не ограничивающих вращениями графовых нейронных сетей для моделирования межатомных потенциалов, ставит вопрос о цене упрощения. Достигнутая сопоставимая точность и скорость — это, безусловно, прогресс, но каждый отчёт о bias в обучении модели — это зеркало общества, отражающее наши собственные предубеждения, закодированные в алгоритмах. Необходимо критически оценить, не упускается ли что-то важное в погоне за вычислительной эффективностью, и какие физические эффекты могут быть недопредставлены в упрощённой модели.
Перспективы развития лежат в плоскости не только повышения точности и скорости, но и в разработке методов интерпретации. Понимание того, как модель приходит к тем или иным предсказаниям, становится столь же важным, как и само предсказание. Необходимо двигаться к созданию моделей, которые не просто имитируют природу, но и позволяют извлекать из неё новые знания. Интерфейс приватности — это форма уважения к пользователю, и аналогичным образом, прозрачность модели — это проявление уважения к науке.
В конечном счёте, задача заключается не в создании всё более сложных и мощных алгоритмов, а в разработке инструментов, которые помогут нам лучше понимать мир вокруг. Прогресс без этики — это ускорение без направления. Будущие исследования должны быть направлены на создание моделей, которые не только точны и эффективны, но и ответственны и прозрачны.
Оригинал статьи: https://arxiv.org/pdf/2601.16195.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Искусственный интеллект, который учится играть: новая платформа для стабильного обучения агентов
- Наука из текста: извлечение знаний из научных публикаций
- Искусственный интеллект: хрупкость визуального мышления
- Сплетение света и времени: аттосекундная спектроскопия на квантовых парах
- Ruyi2: Семейство языковых моделей для эффективного обучения и развертывания
- Самообучающиеся признаки: новый подход к машинному обучению
- Быстрый поиск похожих объектов: GPU-ускорение с IVF-RaBitQ
- Моделирование биомолекул: новый импульс от нейросетей
- Квантовая механика: скрытый детерминизм?
- Агенты SERA: Код, Созданный с Подтверждением
2026-01-25 13:01