Автор: Денис Аветисян
Новый подход к построению межatomных потенциалов на основе машинного обучения позволяет значительно сократить вычислительные затраты и объем памяти без потери точности моделирования.
В статье представлена динамическая функция отсечки для графовых нейронных сетей, снижающая потребление памяти и ускоряющая инференс в молекулярно-динамических симуляциях.
Несмотря на стремительное развитие методов машинного обучения потенциалов взаимодействия атомов, их применение в крупномасштабном моделировании молекулярной динамики ограничено ресурсоёмкостью вычислений и потреблением памяти. В работе ‘Smooth Dynamic Cutoffs for Machine Learning Interatomic Potentials’ предложен новый подход, основанный на использовании динамического радиуса обрезания, позволяющего снизить вычислительные затраты без потери стабильности и точности моделирования. Введение адаптивного обрезания позволяет контролировать разреженность графа взаимодействия, что приводит к снижению потребления памяти в 2.26 раза и ускорению вычислений в 2.04 раза для современных MLIP, таких как MACE, Nequip, Orbv3 и TensorNet. Сможет ли данная методика открыть путь к моделированию материалов и молекул на беспрецедентных временных и пространственных масштабах?
Раскрытие вычислительных барьеров: Новый взгляд на межатомные потенциалы
Метод машинного обучения для создания межатомных потенциалов (MLIP) представляет собой перспективный инструмент для моделирования динамики молекулярных систем, однако его применение часто ограничивается значительными вычислительными затратами. В отличие от традиционных методов, MLIP способны описывать сложные взаимодействия между атомами с высокой точностью, что критически важно для моделирования материалов со сложными свойствами. Тем не менее, обучение и применение этих моделей требует значительных ресурсов, включая время и вычислительную мощность, особенно при моделировании систем, состоящих из большого числа атомов или при проведении длительных молекулярно-динамических симуляций. Это создает серьезные препятствия для изучения масштабных явлений и ограничивает возможности применения MLIP для решения практических задач в материаловедении и химии.
Традиционные методы расчета межмолекулярных взаимодействий, такие как метод фиксированного радиуса обрезки, несмотря на свою вычислительную эффективность, зачастую приводят к потере точности моделирования. Этот подход, ограничивая область рассмотрения взаимодействий определенным расстоянием, игнорирует более слабые, но потенциально важные взаимодействия на больших расстояниях. В результате, моделирование динамики сложных материалов, особенно тех, где дальнодействующие силы играют существенную роль, может давать неверные результаты или требовать значительно больших вычислительных ресурсов для достижения приемлемой точности. Ограничение масштаба моделирования, вызванное необходимостью учета этих дальнодействующих сил, становится серьезным препятствием для изучения свойств реальных материалов и процессов, происходящих в них.
Необходимость в более эффективных межмолекулярных потенциалах, основанных на машинном обучении (MLIP), становится критически важной для моделирования все более сложных материалов и систем. Традиционные методы, хотя и позволяют снизить вычислительные затраты, зачастую компрометируют точность и ограничивают масштаб симуляций. Разработка MLIP, способных к эффективному и точному предсказанию энергии и сил взаимодействия между атомами, открывает возможности для изучения материалов с беспрецедентным уровнем детализации, включая сложные дефекты, динамические процессы и явления, происходящие в экстремальных условиях. Это позволит не только углубить фундаментальное понимание свойств материалов, но и ускорить процесс разработки новых материалов с заданными характеристиками, что имеет огромное значение для различных областей науки и техники, от энергетики до биомедицины.
Динамическая адаптация: Новый принцип отсечения взаимодействий
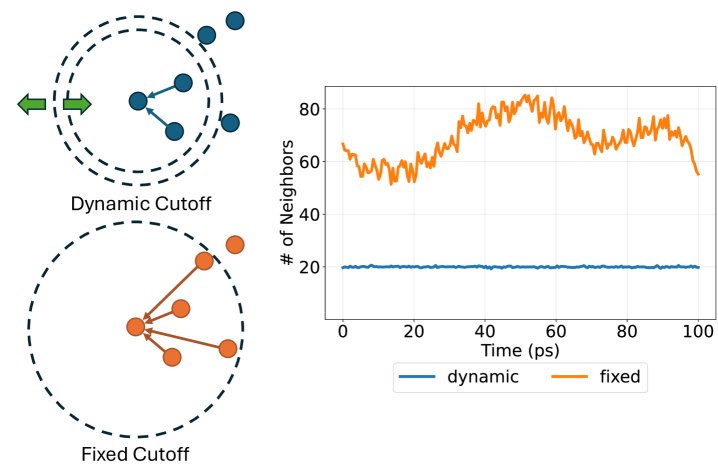
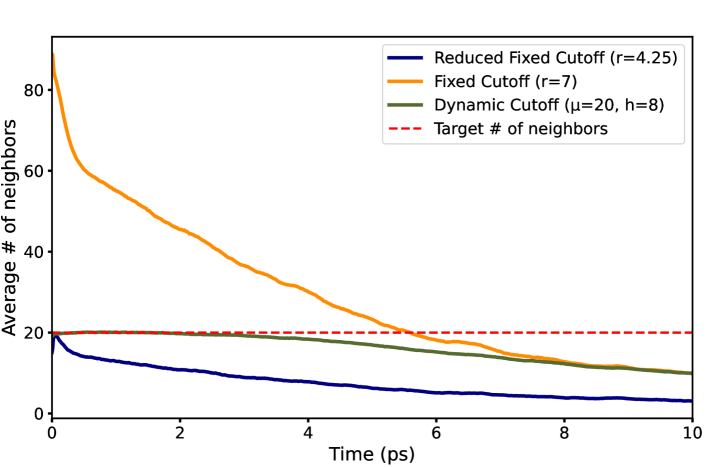
Динамическая функция отсечения адаптирует радиус поиска ближайших соседей, основываясь на локальном окружении каждого атома. Вместо использования фиксированного радиуса для всех атомов, функция анализирует плотность и структуру окружения, увеличивая радиус в разреженных областях и уменьшая его в плотных. Это приводит к построению более разреженного атомного графа, в котором связи устанавливаются только между наиболее значимыми соседями. В результате, количество взаимодействий, которые необходимо вычислить, значительно сокращается, что повышает эффективность моделирования без существенной потери точности.
Метод динамического отсечения использует концепции ранжирования соседей для определения степени влияния удаленных атомов. В основе лежит плавное уменьшение вклада этих соседей посредством применения функций, таких как сигмоидальная функция σ(x) = \frac{1}{1 + e^{-x}} и полиномиальная огибающая. Сигмоидальная функция обеспечивает постепенное снижение веса соседей по мере увеличения расстояния, а полиномиальная огибающая позволяет точно настроить профиль затухания, гарантируя, что только наиболее значимые взаимодействия учитываются в расчетах. Такой подход позволяет эффективно управлять сложностью вычислений, сохраняя при этом необходимую точность моделирования межатомных взаимодействий.
Введение графовой разреженности посредством динамической функции отсечения позволяет существенно снизить вычислительную нагрузку без ущерба для необходимой гладкости потенциальной энергетической поверхности (PES). Уменьшение количества связей в графе, представляющем атомные взаимодействия, приводит к снижению требований к памяти и времени вычислений, особенно при моделировании больших систем. При этом, использование функций плавного затухания, таких как сигмоидальная и полиномиальная огибающая, обеспечивает непрерывность PES, что критически важно для поддержания стабильности и точности молекулярно-динамических симуляций. Уменьшение вычислительной сложности достигается за счет отсечения дальних взаимодействий, вклад которых незначителен, но сохраняется необходимая точность представления PES для обеспечения корректного поведения моделируемой системы.
Экспериментальное подтверждение: Эффективность и точность в действии
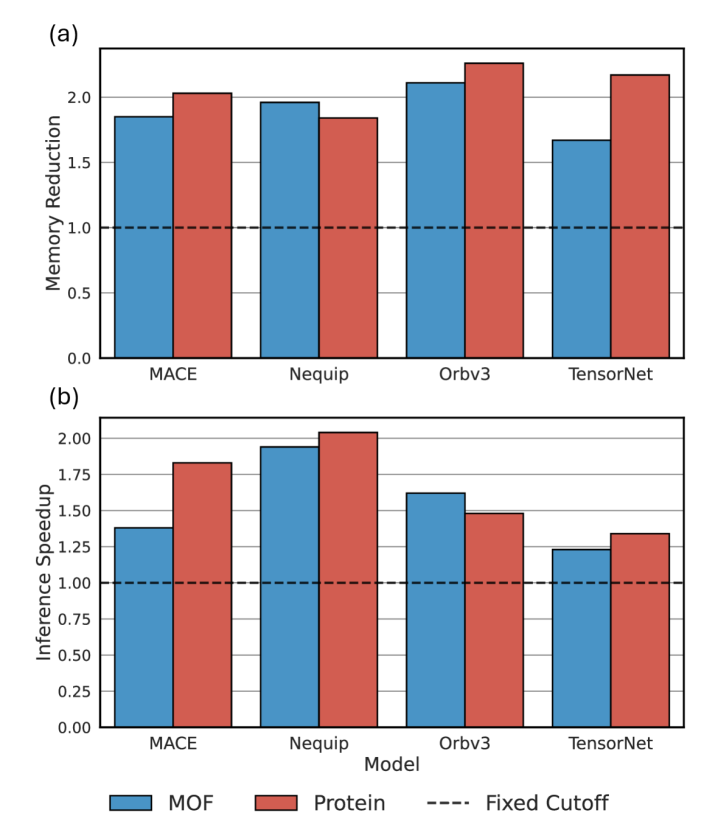
Для всесторонней оценки динамической функции отсечения проводилось тестирование с использованием нескольких моделей машинного обучения потенциалов (MLIP), включая MACE, Nequip, Orbv3 и TensorNet. Выбор данных моделей обусловлен их различной архитектурой и широким применением в задачах моделирования молекулярной динамики. Тестирование проводилось для обеспечения совместимости и эффективности функции отсечения в различных вычислительных окружениях и с разными типами потенциалов, что позволило получить надежные результаты, применимые к широкому спектру задач материаловедения и химии.
В ходе тестирования было зафиксировано значительное снижение времени вычислений и потребления памяти. Максимальное сокращение времени инференса составило 2.04x, а снижение потребления памяти — 2.26x. Данные показатели были получены в результате работы разработанной функции динамического отсечения и отражают существенный прирост производительности при использовании различных моделей машинного обучения потенциальной энергии, таких как MACE, Nequip, Orbv3 и TensorNet.
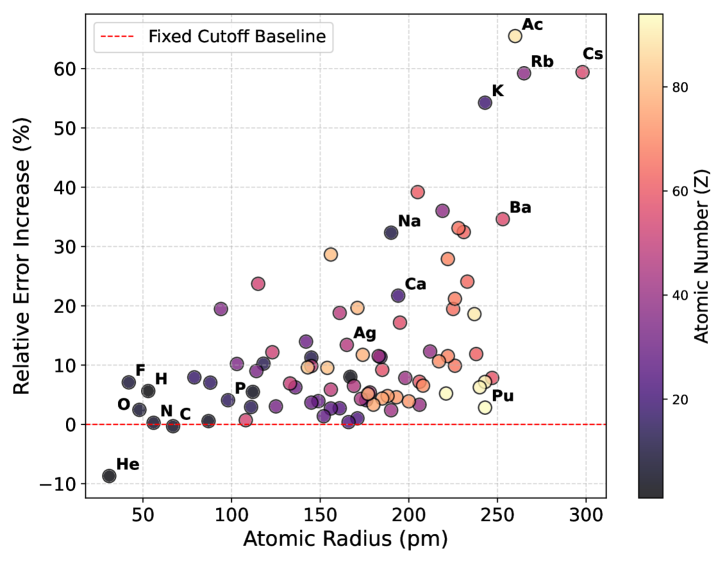
Для подтверждения полученных приростов производительности, функция динамического отсечения была протестирована на стандартных наборах данных MD22 и MatPES. Результаты показали, что снижение времени вычислений и потребления памяти не привело к существенной потере точности. Сравнительный анализ значений средней абсолютной ошибки (MAE) для энергии и сил продемонстрировал их сопоставимость с результатами, полученными без использования функции отсечения, что подтверждает широкую применимость и надежность данного подхода.
За пределами ограничений: Влияние и перспективы нового подхода
Функция динамического отсечения представляет собой существенный прорыв в разработке эффективных и масштабируемых межмолекулярных потенциалов (MLIP). Традиционные методы часто требуют огромных вычислительных ресурсов, ограничивая возможность моделирования сложных материалов и процессов. Новый подход, благодаря адаптивному определению области взаимодействия между атомами, значительно снижает вычислительную сложность без потери точности. Это достигается за счет того, что область расчета потенциальной энергии динамически изменяется в зависимости от расположения атомов, что позволяет исключить ненужные вычисления для удаленных взаимодействий. Такое усовершенствование не только ускоряет моделирование, но и открывает путь к изучению систем, которые ранее были недоступны из-за вычислительных ограничений, что особенно важно для прогнозирования свойств новых материалов и разработки инновационных технологий.
Снижение вычислительных затрат, достигаемое благодаря данной методике, открывает принципиально новые возможности для моделирования материалов, ранее недоступные из-за ограничений ресурсов. Теперь исследователи могут изучать структуры значительно большего размера и сложности, что критически важно для понимания свойств реальных материалов и разработки новых, с заданными характеристиками. Это позволяет ускорить процесс открытия и проектирования материалов для широкого спектра применений, от создания более эффективных аккумуляторов и солнечных панелей до разработки прочных и легких композитных материалов для авиационной и космической промышленности. Возможность моделирования более сложных систем также способствует более точному предсказанию поведения материалов в экстремальных условиях, таких как высокие температуры или давления, что важно для геофизики и материаловедения.
Предстоящие исследования направлены на дальнейшую оптимизацию разработанного метода и расширение области его применения в вычислительной науке о материалах. Ученые планируют усовершенствовать алгоритмы, чтобы добиться еще большей эффективности и масштабируемости, что позволит моделировать системы с беспрецедентной точностью и детализацией. Особое внимание будет уделено адаптации метода для изучения различных классов материалов, включая сложные оксиды, полимеры и биологические молекулы. Это откроет новые возможности для предсказания свойств материалов, разработки инновационных технологий и углубленного понимания фундаментальных принципов, лежащих в основе их поведения. Ожидается, что полученные результаты внесут значительный вклад в области материаловедения, химии и физики, стимулируя дальнейшие научные открытия и технологические прорывы.
Исследование демонстрирует подход к оптимизации вычислений в молекулярной динамике, где адаптивное определение окрестности атома позволяет существенно снизить вычислительные затраты и объем потребляемой памяти. Этот метод, по сути, переосмысливает понятие «ограничения» в расчетах, фокусируясь на действительно значимых взаимодействиях. Кен Томпсон однажды заметил: «Программы должны быть достаточно маленькими, чтобы их можно было понять». Подобно этому, предложенная функция отсечения стремится к минимально необходимому объему вычислений, сохраняя при этом стабильность модели. Упрощение системы, отказ от избыточных параметров — это путь к более эффективному и понятному решению, особенно в сложных областях, таких как машинное обучение межатомных потенциалов.
Куда Далее?
Представленная работа, по сути, лишь аккуратный обход одного из фундаментальных ограничений — экспоненциальной сложности расчётов в многочастичных системах. Уменьшение числа учитываемых соседей — не решение, а временная отсрочка неизбежного столкновения с необходимостью принципиально новых подходов. Ускорение и снижение потребления памяти — полезные побочные эффекты, но истинный вызов заключается в создании потенциалов, которые не нуждатся в жёстких ограничениях ради вычислительной эффективности.
Будущие исследования, вероятно, будут сосредоточены на преодолении самой концепции фиксированного радиуса обрезания. Возможно, стоит взглянуть в сторону потенциалов, учитывающих не только геометрическую близость атомов, но и их квантово-механическое взаимодействие, пусть и в приближённом виде. Или, возможно, стоит признать, что истинная симуляция сложных материалов требует принципиально иных вычислительных архитектур — тех, которые способны эффективно работать с разреженными графами и нелинейными зависимостями.
В конечном счёте, данная работа напоминает о том, что хаос — не враг, а зеркало архитектуры, которое отражает скрытые связи. Обрезание графа — это не упрощение, а искажение. Истинное понимание требует не игнорирования сложности, а её освоения.
Оригинал статьи: https://arxiv.org/pdf/2601.21147.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Искусственный интеллект, который учится играть: новая платформа для стабильного обучения агентов
- Вероятностный компьютер на фотонных чипах: новая эра вычислений
- Наука из текста: извлечение знаний из научных публикаций
- Моделирование биомолекул: новый импульс от нейросетей
- Биомолекулярные связи: новый тест для искусственного интеллекта
- Робот-исследователь: новый подход к автономной навигации
- Ruyi2: Семейство языковых моделей для эффективного обучения и развертывания
- Сплетение света и времени: аттосекундная спектроскопия на квантовых парах
- Самообучающиеся признаки: новый подход к машинному обучению
- Искусственный интеллект: хрупкость визуального мышления
2026-02-02 01:13