Автор: Денис Аветисян
Исследователи разработали метод предсказания распределения белков в тканях, используя данные о пространственной транскриптомике и другие омиксные данные.
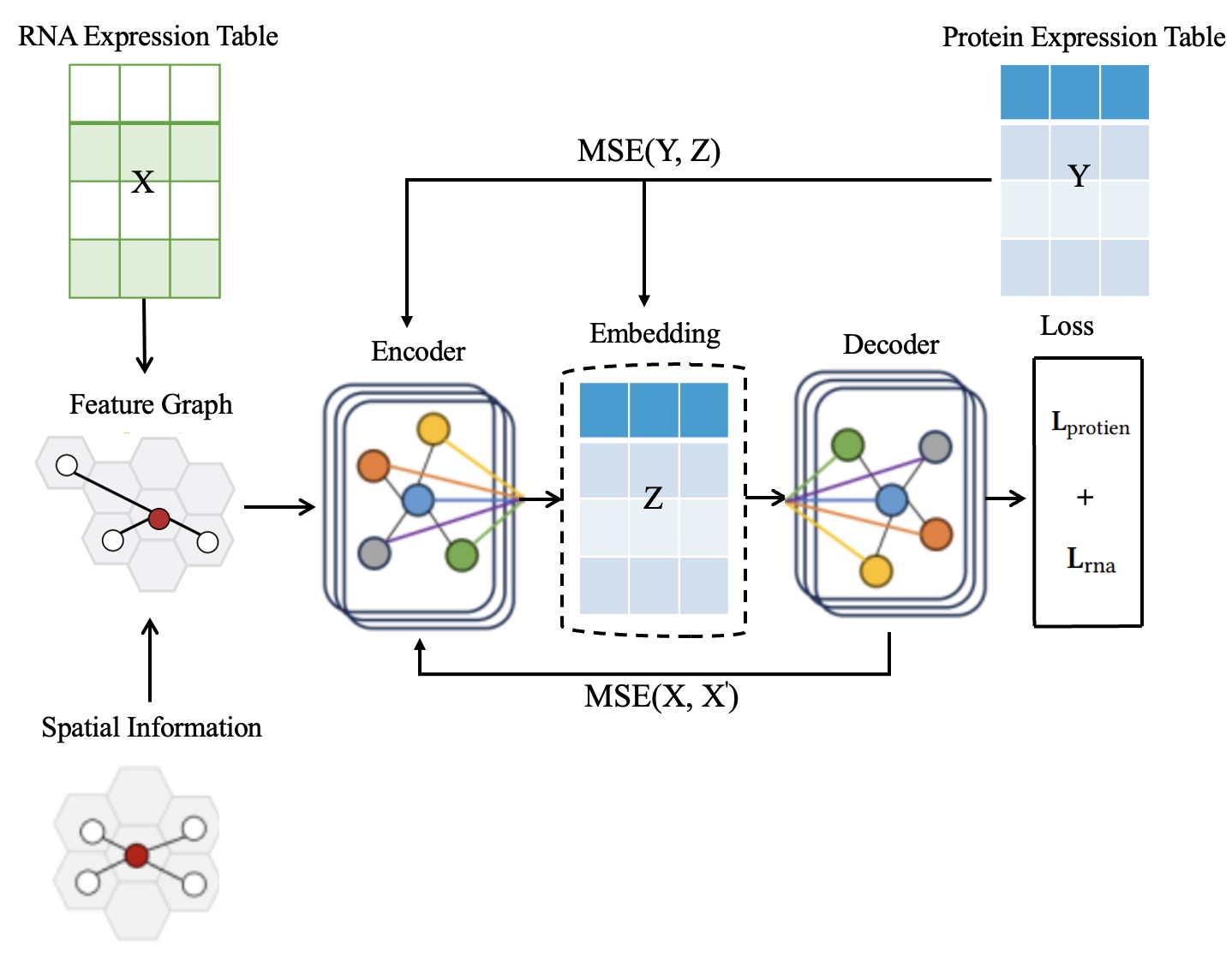
Представлена платформа STProtein, использующая графовые нейронные сети для точного прогнозирования пространственного распределения белков на основе данных мультиомики.
Несмотря на растущий объем пространственных мультиомиксных данных, существенный дисбаланс между доступностью пространственной транскриптомики и протеомики сдерживает прогресс в изучении тканевой биологии. В данной работе представлена платформа ‘STProtein: predicting spatial protein expression from multi-omics data’, использующая графовые нейронные сети и многозадачное обучение для точного предсказания пространственного распределения белков на основе более доступных мультиомиксных данных. Разработанный подход позволяет эффективно преодолеть дефицит пространственной протеомики, открывая новые возможности для интеграции мультиомиксных данных и изучения ранее скрытых закономерностей в тканях. Позволит ли STProtein выявить новые биомаркеры и углубить наше понимание сложных биологических процессов?
Пространственная Биология: Вызовы Анализа Данных
Традиционные методы биологических исследований зачастую не позволяют в полной мере оценить сложность организации тканей, что приводит к потере важной пространственной информации. Исторически анализ биологических образцов сводился к усредненным измерениям, например, изучению всей ткани как единого целого, что не учитывало гетерогенность клеточного состава и взаимодействие между клетками в их естественном окружении. Это подобно попытке понять картину, рассматривая лишь размытый общий вид, вместо того, чтобы видеть детали каждого мазка и нюанс расположения элементов. В результате, критически важные сигналы, определяемые именно расположением клеток и молекул внутри ткани, оказываются упущенными, что ограничивает возможности понимания процессов развития, функционирования и возникновения заболеваний. Современные исследования все больше внимания уделяют необходимости сохранения и анализа этой пространственной информации для получения более полного и точного представления о биологических системах.
Развитие пространственной омики привело к генерации огромных массивов данных, однако их интеграция представляет собой значительную проблему. Современные технологии, такие как пространственная транскриптомика и протеомика, позволяют картировать молекулярные профили клеток в контексте их местоположения в ткани, создавая многомерные наборы данных. Сложность заключается в объединении этих разнородных слоев информации — от данных о генах и белках до морфологических характеристик — в единую, целостную картину. Необходимы новые вычислительные методы и алгоритмы для эффективной обработки, анализа и интерпретации этих данных, чтобы выявить закономерности и связи, скрытые в пространственной организации тканей. Успешное решение этой задачи откроет возможности для более глубокого понимания биологических процессов, диагностики заболеваний и разработки новых терапевтических стратегий.
Значительная проблема современной биологии заключается в расшифровке так называемого “темного вещества” биологических данных — неаннотированных областей тканей, которые зачастую остаются без внимания при традиционном анализе. Эти регионы, кажущиеся пустыми или несущественными, могут содержать ключевую информацию о межклеточных взаимодействиях, регуляции генов и даже о возникновении заболеваний. Современные технологии пространной омики позволяют картировать эти области с беспрецедентной точностью, однако интерпретация этих данных требует разработки новых алгоритмов и подходов к анализу, способных выявить скрытые закономерности и биологическую значимость “темного вещества”. Понимание этих ранее неизученных участков тканей может радикально изменить представления о функционировании организма и открыть новые возможности для диагностики и лечения различных патологий.

STProtein: Рациональная Основа Пространственного Предсказания
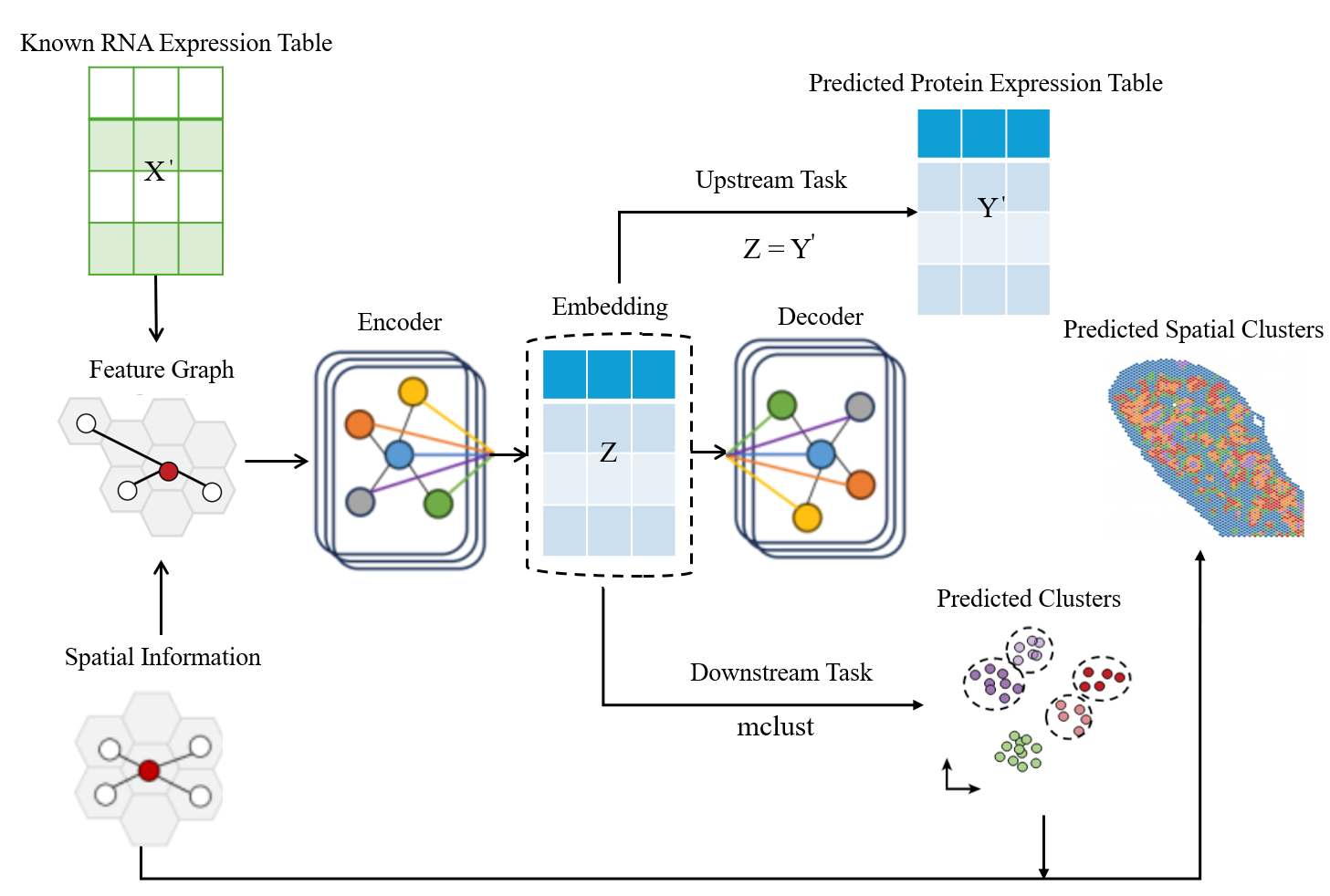
STProtein — это новый программный комплекс, предназначенный для предсказания пространственного распределения экспрессии белков на основе данных мультиомиксных исследований. Фреймворк позволяет анализировать данные, полученные различными методами, включая геномику, транскриптомику и протеомику, для построения карт пространственного распределения белков в тканях. Это достигается путем интеграции данных различных типов и применения алгоритмов машинного обучения для прогнозирования уровня экспрессии белков в каждой точке ткани, что позволяет получить детальное представление о пространственной организации протеома.
STProtein использует граф на основе k-ближайших соседей (KNN) для представления пространственных взаимосвязей между различными точками ткани. В данном подходе, каждый узел графа соответствует конкретному пространственному положению, а ребра соединяют узлы, представляющие близлежащие позиции, отобранные на основе расстояния и сходства признаков. Конструкция KNN-графа позволяет эффективно моделировать структуру ткани, учитывая локальную близость и взаимосвязь между клетками или областями, что критически важно для точного предсказания пространственного распределения белков. Параметр k определяет количество ближайших соседей, используемых для установления связей, и его оптимальное значение подбирается для достижения наилучшей производительности модели.
Метод STProtein использует графовые нейронные сети (GNN) для выявления сложных закономерностей в пространственных данных экспрессии белков. GNN позволяют анализировать взаимосвязи между различными участками ткани, представленными как узлы графа, и учитывать их окружение как ребра. Это позволяет моделировать нелинейные зависимости и учитывать контекстную информацию, недоступную при традиционных методах анализа данных. Обучение GNN происходит на основе графа, построенного с использованием KNN, что позволяет эффективно использовать информацию о пространственной близости и структуре ткани для прогнозирования экспрессии белков в каждой точке.

Проверка Точности и Надежности Прогнозов
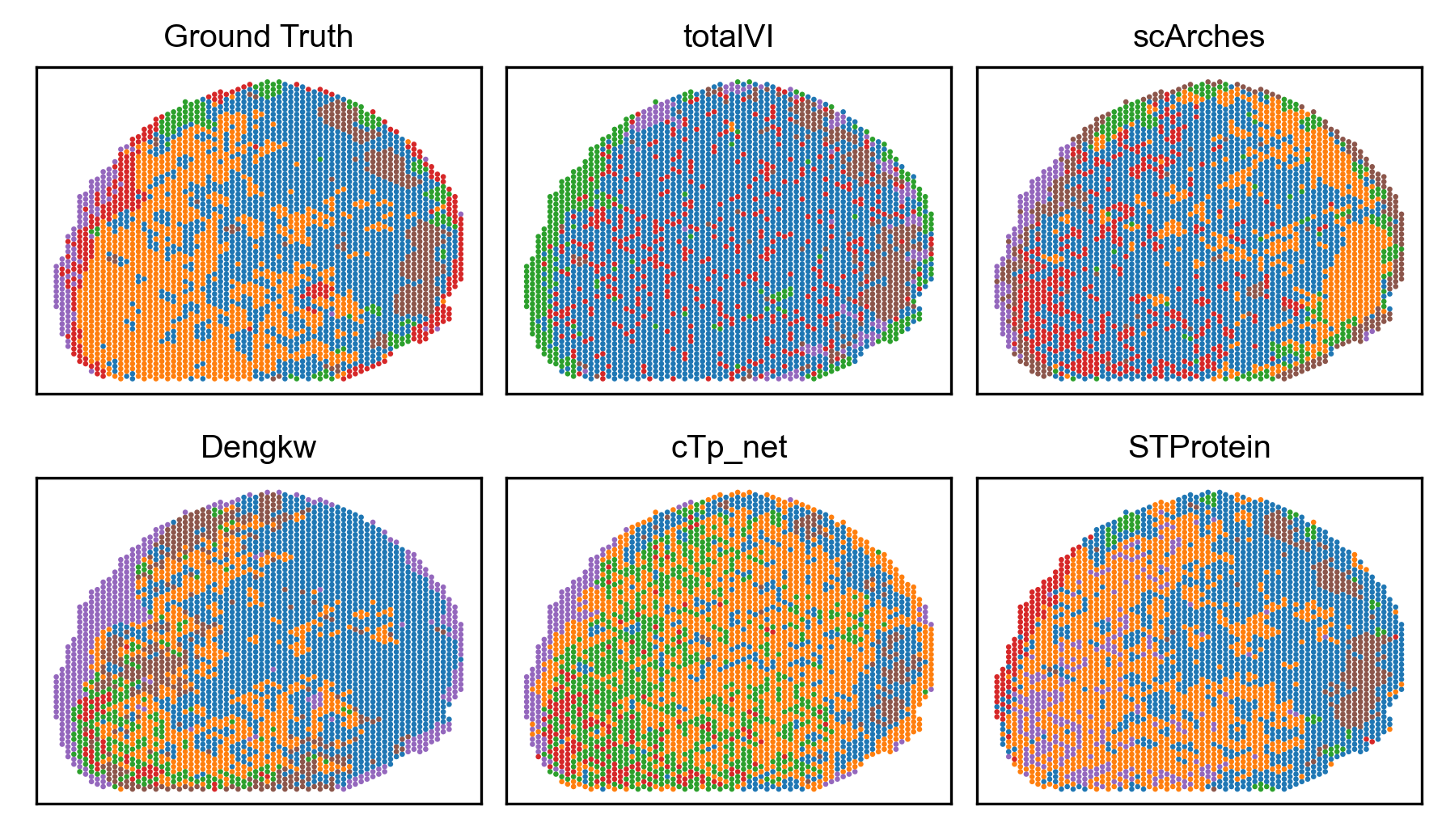
Эффективность STProtein оценивалась с использованием среднеквадратичной ошибки (RMSE) в качестве ключевого показателя, демонстрируя превосходную точность предсказаний по сравнению с альтернативными методами. На датасетах Mouse Spleen, Mouse Thymus и Human Lymph Node, STProtein показал RMSE на 0.04, 0.07 и 0.17 единицы ниже, чем у других сравниваемых методов, что свидетельствует о более высокой точности прогнозирования в данных исследованиях.
В основе STProtein лежит использование проверенных инструментов предварительной обработки данных, таких как Seurat, что обеспечивает надежную подготовку данных для последующего анализа. Для повышения эффективности и снижения вычислительной сложности, в рамках системы применяются методы понижения размерности, в частности, анализ главных компонент (PCA) и алгоритм UMAP. Эти методы позволяют сократить количество признаков в данных, сохраняя при этом наиболее важную информацию, что способствует более быстрой и точной обработке данных пространственной транскриптомики.
В рамках STProtein используются методы кластеризации, демонстрирующие наивысшие значения среди сравниваемых подходов по ряду метрик оценки качества. В частности, при анализе данных достигаются лучшие показатели по метрикам Normalized Mutual Information (NMI), Adjusted Mutual Information (AMI), Fowlkes-Mallows Index (FMI), Adjusted Rand Index (ARI), Homogeneity, V-measure и F1-Score. Данные результаты подтверждают эффективность алгоритмов кластеризации, реализованных в STProtein, для точной идентификации и группировки клеток в пространственных данных транскриптома.
Влияние и Перспективы Развития
Метод STProtein эффективно решает проблему дисбаланса данных, часто встречающуюся при анализе пространственной мультиомики. В традиционных подходах неравномерное распределение данных между различными типами клеток или участками ткани может приводить к смещенным результатам и ненадежным прогнозам. STProtein, используя специализированные алгоритмы, компенсирует этот дисбаланс, обеспечивая более сбалансированную оценку значимости различных факторов. Это позволяет получать более точные и надежные предсказания, что особенно важно при изучении сложных биологических процессов, таких как развитие заболеваний. В результате, STProtein открывает возможности для более глубокого и объективного анализа пространственных данных, повышая достоверность научных выводов и способствуя разработке новых методов диагностики и лечения.
Метод STProtein демонстрирует значительный потенциал в различных областях биомедицинских исследований. Он позволяет глубже понять механизмы развития заболеваний, отслеживая изменения в пространственной организации клеток и их молекулярных профилях. Это, в свою очередь, открывает возможности для выявления новых терапевтических мишеней, направленных на конкретные молекулярные пути, вовлеченные в патогенез. Кроме того, благодаря возможности точного определения клеточных состояний и их пространственного расположения, STProtein способствует разработке персонализированных стратегий лечения, учитывающих индивидуальные особенности пациента и характеристики его заболевания. В перспективе, этот подход может значительно повысить эффективность терапии и улучшить прогнозы для пациентов с различными заболеваниями.
Перспективные исследования сосредоточены на расширении возможностей STProtein за счет интеграции дополнительных слоев омиксных данных, что позволит получить более полное представление о биологических процессах. Параллельно изучается применение методов переноса обучения, таких как scArches и totalVI, для повышения точности прогнозов и адаптации модели к различным типам данных. Значительным достижением, обусловленным применением STProtein, стало обнаружение и аннотация ранее неизвестных подтипов макрофагов (MZMΦ и MMMΦ) в селезенке мыши. Это открытие существенно расширяет существующие знания о составе тканей и открывает новые перспективы для изучения иммунных реакций и патогенеза заболеваний, позволяя глубже понять сложность клеточных взаимодействий в тканях.
Представленная работа демонстрирует стремление к математической чистоте в анализе биологических данных. Разработанный фреймворк STProtein, использующий графовые нейронные сети, направлен на точную предсказацию пространственного выражения белков на основе мультиомиксных данных. Это особенно важно, учитывая ограниченность данных пространственной протеомики. Как отмечал Дональд Дэвис: «Простота — это высшая степень утонченности». Этот принцип находит отражение в элегантном подходе к интеграции данных и построении модели, позволяющей получить ценные сведения о биологии тканей, минимизируя избыточность и фокусируясь на корректности алгоритма.
Что дальше?
Представленная работа, безусловно, демонстрирует элегантность подхода к интеграции разнородных омиксных данных для предсказания пространственного распределения белков. Однако, истинная строгость научного метода требует признания границ применимости. Модель, как и любой инструмент, опирается на предположения о природе биологических систем. Вопрос о том, насколько адекватно графовые нейронные сети отражают реальные взаимодействия в ткани, остаётся открытым и требует дальнейшего изучения. Простое увеличение объёма обучающей выборки не решит фундаментальную проблему — необходимость формального доказательства корректности предсказаний.
Наиболее перспективным направлением представляется разработка методов верификации предсказанных белковых карт. Экспериментальная проверка предсказаний, хотя и трудоёмка, необходима для оценки надёжности модели и выявления её слабых мест. Кроме того, следует уделить внимание разработке метрик, способных оценивать не только точность предсказаний, но и их биологическую значимость. Простое соответствие данным — недостаточный критерий, требуется понимание того, как предсказанные изменения в распределении белков влияют на функцию ткани.
В конечном счёте, задача предсказания пространственного распределения белков — это лишь один шаг на пути к созданию полной, математически строгой модели ткани. Истинная красота алгоритма проявится не в достижении высокой точности на тестовых данных, а в способности предсказывать поведение ткани в условиях, отличных от тех, что были использованы при обучении. Иначе говоря, необходима универсальность, а не просто адаптация к конкретным условиям.
Оригинал статьи: https://arxiv.org/pdf/2602.05811.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Искусственный интеллект, который учится играть: новая платформа для стабильного обучения агентов
- Автопилот нового поколения: Единая модель для понимания, планирования и предвидения
- Видеть детали: новый подход к мультимодальному восприятию
- Сердце под контролем ИИ: новый подход к диагностике
- Эхо разума: как итеративные модели учатся в цикле.
- Квантовые вычисления: линейная алгебра на службе симуляции
- Восстановление электронной структуры материалов с помощью машинного обучения
- Разум как отражение: новая архитектура интеллекта
- Квантовая электродинамика и сильные корреляции: новый взгляд на взаимодействие света и материи
- Квантовые вычисления: Новый взгляд на оценку ресурсов
2026-02-08 06:56