Автор: Денис Аветисян
Исследователи разработали гибридный квантово-классический алгоритм, позволяющий более точно моделировать поведение электронов в сложных химических системах.
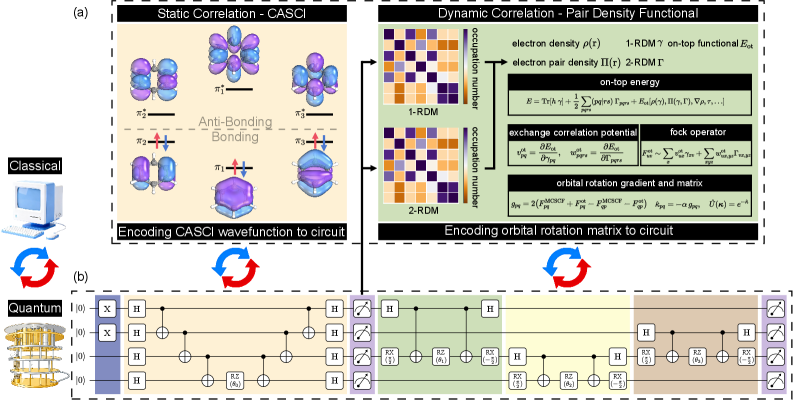
В статье представлен метод VQE-MC-PDFT, объединяющий вариационный квантовый эйнзольвер и многоконфигурационную теорию плотности пар, для высокоточного расчета основного и возбужденных состояний молекул.
Точное описание сильной электронной корреляции в сложных химических системах остается сложной задачей в вычислительной химии, особенно с учетом ограничений существующих квантовых алгоритмов. В данной работе, посвященной ‘Multiconfiguration Pair-Density Functional Theory Calculations of Ground and Excited States of Complex Chemical Systems with Quantum Computers’, предложен гибридный подход, сочетающий вариационный квантовый эйнзольвер (VQE) с многоконфигурационной теорией плотности пар (MC-PDFT) для эффективного разделения эффектов корреляции. Такой подход позволяет снизить требования к квантовым ресурсам, сохраняя при этом физическую строгость и достигая химической точности в расчетах, например, воспроизводя длины связей в молекуле C_2 с ошибкой 0.006 Å. Может ли подобное разделение типов корреляции стать практическим путем к надежным предсказаниям на перспективных квантовых вычислительных платформах?
Предел Классических Расчетов: Эпоха Неизбежных Ошибок
Многочастичные квантовые системы представляют собой серьезную проблему для вычислительной химии, поскольку для их точного описания требуются вычислительные ресурсы, растущие экспоненциально с увеличением числа частиц. Это связано с тем, что волновой функцией, определяющей состояние такой системы, необходимо описать корреляции между всеми частицами, что приводит к огромному объему вычислений. Например, для описания всего лишь нескольких электронов в молекуле необходимо учитывать все возможные комбинации их взаимного расположения и взаимодействия. O(N!), где N — количество частиц, приблизительно отражает рост вычислительной сложности с увеличением размера системы. Такой экспоненциальный рост делает точное моделирование даже относительно небольших молекул непосильной задачей для классических компьютеров, стимулируя поиск новых, более эффективных алгоритмов и вычислительных подходов.
Традиционные методы вычислительной химии, такие как метод Хартри-Фока и теория функционала плотности, несмотря на широкое распространение, сталкиваются с серьезными трудностями при моделировании систем с сильной корреляцией электронов. В таких системах взаимодействие между электронами становится доминирующим, и приближения, лежащие в основе этих методов, приводят к существенным погрешностям в предсказаниях свойств молекул и материалов. Например, при описании переходных металлов или систем с частично заполненными d-оболочками, стандартные функционалы плотности часто не способны адекватно учесть электронную корреляцию, что приводит к неверным оценкам энергии, геометрии и спектральных характеристик. Это ограничивает применимость этих методов к широкому кругу важных химических и физических задач, требуя разработки более совершенных подходов для точного моделирования сильно коррелированных систем.
Методы, такие как UCCSD (полная конфигурационная корреляция с единичными и двойными возбуждениями) и CASSCF (полная конфигурационная корреляция с включением активных электронов), демонстрируют повышенную точность при моделировании электронных структур сложных молекул, однако их вычислительная стоимость резко возрастает с увеличением размера молекулы. Проблема заключается в экспоненциальном масштабировании «активного пространства» — набора орбиталей и электронов, участвующих в корреляционных расчетах. Каждый добавленный электрон или орбиталь увеличивает размер матрицы, описывающей взаимодействие между ними, экспоненциально увеличивая требуемые ресурсы памяти и процессорного времени. В результате, применение этих методов становится практически невозможным для молекул, содержащих даже относительно небольшое количество электронов, ограничивая их использование в исследованиях более крупных и сложных систем, представляющих интерес для химии и материаловедения.
В связи с ограничениями традиционных методов электронных структур, разработка гибридных квантово-классических алгоритмов становится необходимостью для моделирования сложных молекулярных систем. Эти алгоритмы стремятся использовать преимущества как квантовых, так и классических вычислений: квантовые компьютеры выполняют задачи, непосильные для классических машин, например, моделирование сильной корреляции электронов, в то время как классические компьютеры управляют процессом и обрабатывают большие объемы данных. Такой подход позволяет преодолеть экспоненциальный рост вычислительных затрат, характерный для точных методов, и открывает возможности для изучения молекул и материалов, которые ранее были недоступны для детального теоретического анализа. Перспективные алгоритмы, такие как вариационный квантовый эйнштейнский решатель (VQE) и квантовый фазовый оценщик (QPE), демонстрируют потенциал для решения задач в химии и материаловедении с беспрецедентной точностью и эффективностью.
Синергия Гибридных Подходов: VQE-MC-PDFT
Вариационный квантовый решатель (VQE) представляет собой алгоритм, предназначенный для нахождения приближенных собственных состояний основного уровня на квантовых устройствах ближнего будущего. В отличие от методов, требующих больших квантовых ресурсов, VQE использует гибридный классическо-квантовый подход, в котором квантовый компьютер используется для оценки энергии предложенного волнового пакета, а классический компьютер оптимизирует параметры этого пакета для минимизации энергии. Этот процесс итеративно повторяется до достижения сходимости, что позволяет находить приближенные решения для молекулярных систем и материалов, которые сложно моделировать классическими методами. Алгоритм VQE особенно полезен для систем, где точное решение уравнения Шредингера невозможно из-за экспоненциального роста вычислительных затрат с увеличением числа частиц.
Точность алгоритма VQE напрямую зависит от выбора анзаца — параметризованного квантового состояния, используемого для представления искомого основного состояния системы. Ограниченный анзац может не охватить все важные корреляции между электронами, что приводит к неточному результату. Кроме того, сложность квантовой схемы, необходимой для реализации выбранного анзаца, возрастает с увеличением числа кубитов и глубины цепи, что может превысить возможности современных квантовых устройств из-за ограничений по когерентности и шуму. Повышение точности VQE часто требует компромисса между сложностью анзаца и возможностью его реализации на доступном квантовом оборудовании.
Интеграция Variational Quantum Eigensolver (VQE) с Multiconfiguration Pair-Density Functional Theory (MC-PDFT) представляет собой перспективный подход к повышению точности и снижению вычислительных затрат при решении задач квантовой химии. MC-PDFT использует Reduced Density Matrix (RDM) и on-top pair density для эффективного описания электронных корреляций, что позволяет сократить сложность квантовой схемы, необходимой для VQE. Вместо явного моделирования всех возбуждений, MC-PDFT предоставляет точное описание ключевых корреляционных эффектов, которые затем учитываются в вариационном принципе VQE. Такой гибридный подход позволяет VQE достигать более высокой точности с меньшим количеством кубитов и глубиной квантовой цепи, что особенно важно для реализации на современных и перспективных квантовых устройствах.
Многоконфигурационная теория функционала плотности пар (MC-PDFT) использует редуцированную матрицу плотности (RDM) и плотность пар “on-top” для эффективного описания эффектов электронной корреляции. RDM, представляющая собой матрицу плотности, ограниченную определенным набором детерминантов, позволяет отбросить ненужные конфигурации, снижая вычислительную сложность. Плотность пар “on-top” описывает корреляцию между электронами, находящимися в одной пространственной области, и позволяет точно оценить энергию корреляции. В сочетании с вариационным принципом, лежащим в основе Variational Quantum Eigensolver (VQE), MC-PDFT предоставляет более точный и эффективный подход к решению задач квантовой химии, особенно для систем, требующих учета сильных корреляционных эффектов. Использование RDM и плотности пар “on-top” позволяет сократить размер базисного набора и упростить вычисление энергии корреляции, что критически важно для реализации на квантовых устройствах.
Применение стратегий смягчения ошибок в рамках алгоритмической схемы VQE-MC-PDFT является критически важным для получения достоверных результатов на современных квантовых устройствах. Метод VQE-MC-PDFT, сочетающий вариационный квантовый эвристический алгоритм (VQE), метод Монте-Карло (MC) и функционал плотности (PDFT), требует высокой точности квантовых вычислений для корректного моделирования электронных структур. Из-за подверженности промежуточных квантовых устройств (NISQ) шумам и ошибкам, без использования методов смягчения ошибок, таких как экстраполяция к нулевому шуму (ZNE) и подгонка Клиффорда (CF), результаты вычислений оказываются недостоверными и не отражают реальные физические свойства исследуемых систем. Внедрение этих стратегий позволяет уменьшить влияние ошибок и получить более точные оценки энергии и других квантово-механических характеристик, что необходимо для практического применения квантовых вычислений в материаловедении и химии.
Оценка и Перспективы: Эпоха Квантовой Химии
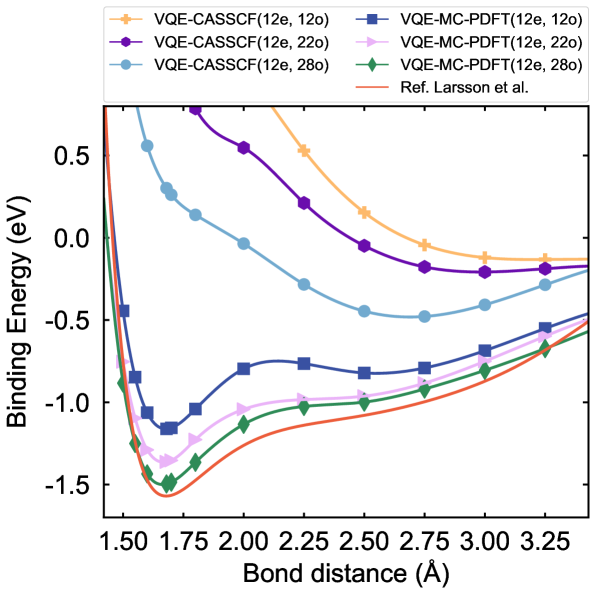
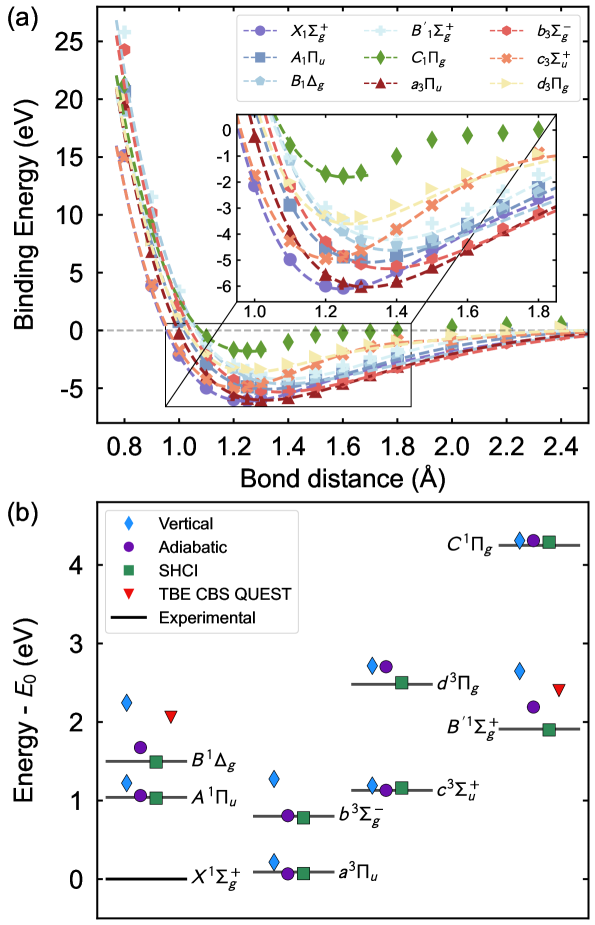
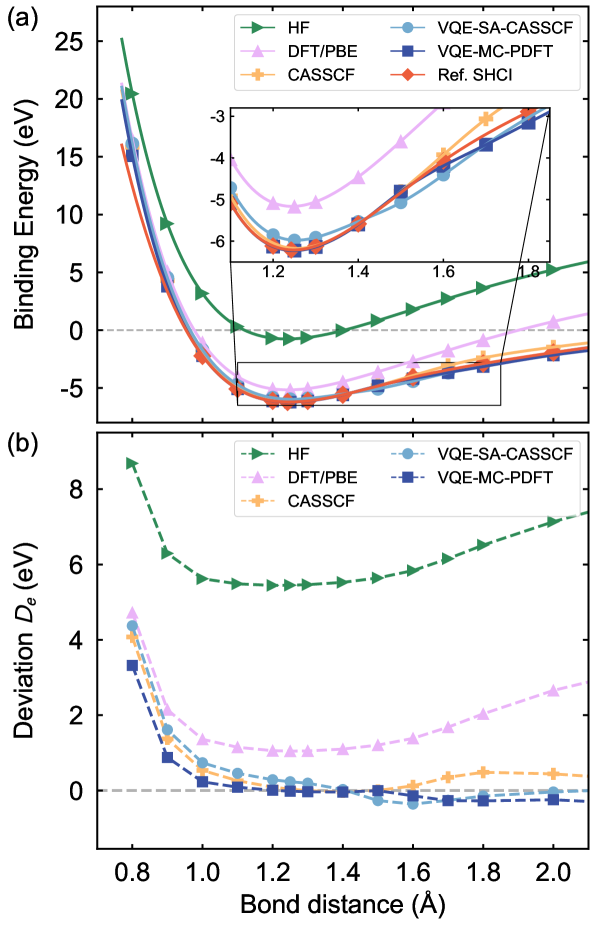
Метод VQE-MC-PDFT успешно протестирован на эталонных молекулах, таких как C2 и бензол, продемонстрировав высокую точность предсказания энергии основного состояния. Исследования показали, что данный подход способен надежно воспроизводить известные результаты для этих относительно простых систем, что является важным шагом к применению метода для более сложных молекулярных структур. Точность, достигнутая в расчетах энергии основного состояния для C2 и бензола, подтверждает эффективность комбинирования вариационного квантового эвристического алгоритма (VQE) с методом Монте-Карло и функцией плотности, открывая перспективы для изучения молекул и материалов, ранее недоступных для детального теоретического анализа.
Исследования показали, что предложенный метод VQE-MC-PDFT успешно применяется не только к простым молекулам, но и к более сложным системам, таким как диатомная молекула хрома (Cr2). Вычисление энергии связи для Cr2 дало результат в -0.465 эВ, что является конкурентоспособным показателем, сопоставимым с результатами, полученными с использованием эталонных методов теоретического расчета. Данное достижение демонстрирует потенциал метода для изучения систем с сильными электронными корреляциями, которые представляют значительный интерес для материаловедения и химии, и открывает возможности для точного моделирования сложных молекулярных взаимодействий.
Применительно к бензолу, разработанный метод демонстрирует высокую точность предсказания энергий возбужденных состояний, достигая средней абсолютной ошибки (MAE) в 0.048 эВ. Этот показатель сопоставим с результатами, полученными с использованием наиболее передовых теоретических подходов, что свидетельствует о надежности и эффективности метода в моделировании сложных органических молекул. Достигнутая точность открывает перспективы для детального изучения спектроскопических свойств бензола и его производных, а также для разработки новых материалов с заданными оптическими характеристиками. Полученные результаты подчеркивают значительный прогресс в области квантово-химических расчетов и приближают возможность моделирования еще более сложных молекулярных систем.
Метод, использующий фрагментарную реконструкцию, демонстрирует впечатляющую масштабируемость, требуя до 84 кубитов для проведения сложных вычислений. Такой подход позволяет преодолеть ограничения, накладываемые текущими возможностями кванкольного оборудования, поскольку позволяет разбить большую молекулярную систему на более мелкие, управляемые фрагменты. Каждый фрагмент рассчитывается отдельно, а затем результаты объединяются для получения общей картины. Это не только снижает потребность в большом количестве кубитов, но и повышает точность расчетов, делая возможным изучение молекулярных систем, которые ранее были недоступны для моделирования. Использование фрагментарной реконструкции открывает перспективы для разработки новых материалов и химических соединений, требующих точного моделирования сложных молекулярных структур.
Данный гибридный подход открывает принципиально новые возможности для изучения молекулярных систем, расчеты для которых ранее были недоступны из-за вычислительных ограничений. Комбинируя преимущества квантовых и классических вычислений, методика позволяет моделировать сложные молекулы с высокой точностью, что имеет решающее значение для проектирования новых материалов и химических соединений. Благодаря возможности преодолевать ограничения, связанные с текущими аппаратными ресурсами, становится возможным предсказывать свойства веществ с заданными характеристиками, оптимизировать их структуру и, в конечном итоге, ускорить процесс открытия инновационных материалов с улучшенными свойствами, например, для использования в катализе, солнечной энергетике или создании новых лекарственных препаратов. Данная методология представляет собой значительный шаг вперед в области вычислительной химии и материаловедения, потенциально революционизирующий процесс разработки новых технологий.
Дальнейшие исследования направлены на оптимизацию квантовых схем и разработку более надежных стратегий смягчения ошибок, что позволит полностью раскрыть потенциал данного мощного метода. Улучшение эффективности схем позволит обрабатывать более сложные молекулярные системы, требующие меньшего количества кубитов и времени вычислений. Параллельно, разработка усовершенствованных методов смягчения ошибок, компенсирующих влияние декогеренции и других неточностей, критически важна для получения достоверных результатов. Эти усилия позволят преодолеть текущие ограничения квантового оборудования и расширить возможности моделирования молекул, что откроет новые перспективы в материаловедении и химическом дизайне, позволяя предсказывать свойства и поведение веществ с беспрецедентной точностью.
Исследование, представленное в данной работе, демонстрирует стремление к преодолению фундаментальных ограничений в моделировании сложных химических систем. Авторы предлагают гибридный алгоритм, объединяющий возможности квантовых вычислений и классической теории функционала плотности. Этот подход, как и любое усложнение системы, несет в себе потенциал для новых сбоев, новых зависимостей. Как заметил Дональд Кнут: «Преждевременная оптимизация - корень всех зол». В данном случае, стремление к точности и эффективности требует тщательного учета возникающих компромиссов, ведь каждая архитектурная деталь - это пророчество о будущей ошибке. Игнорирование этого факта обрекает систему на неминуемые проблемы, какими бы элегантными ни казались ее начальные принципы.
Что Дальше?
Представленный здесь алгоритм, VQE-MC-PDFT, подобен тщательно взращенному растению, но даже самое крепкое дерево имеет свои пределы. Попытка обернуть сложность многоконфигурационного подхода в рамки вариационного принципа - это не победа над энтропией, а лишь её временное умиротворение. Неизбежно возникнет потребность в более изящных функциях корреляции, способных улавливать тончайшие взаимодействия между электронами, не требуя экспоненциального увеличения вычислительных затрат. Иначе этот паттерн выродится через три релиза, увязнув в проблемах масштабируемости.
Особое внимание следует уделить ошибкам, неизбежно возникающим в квантовых вычислениях. Методы смягчения ошибок - это не панацея, а лишь отсрочка неизбежного. Истинный прогресс заключается в разработке алгоритмов, устойчивых к шуму, которые используют эти ошибки как источник информации, а не как препятствие. В каждом кроне скрыт страх перед хаосом, и лишь адаптация к нему позволит этой ветви прорасти.
В конечном счете, успех этого направления зависит не от совершенства алгоритмов, а от способности исследователей взглянуть на молекулярную структуру как на сложную экосистему, а не на набор атомов и электронов. Надежда на идеальную архитектуру - это форма отрицания энтропии. Будущее квантовой химии - это не в создании идеальных моделей, а в понимании и принятии её присущей неопределенности.
Оригинал статьи: https://arxiv.org/pdf/2602.10435.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Искусственный интеллект, который учится играть: новая платформа для стабильного обучения агентов
- Вероятностный компьютер на фотонных чипах: новая эра вычислений
- Моделирование биомолекул: новый импульс от нейросетей
- Наука из текста: извлечение знаний из научных публикаций
- Искусственный интеллект: хрупкость визуального мышления
- Наука на Автопилоте: Система для Самостоятельных Исследований
- Сплетение света и времени: аттосекундная спектроскопия на квантовых парах
- Ruyi2: Семейство языковых моделей для эффективного обучения и развертывания
- Биомолекулярные связи: новый тест для искусственного интеллекта
- Робот-исследователь: новый подход к автономной навигации
2026-02-12 10:33