Автор: Денис Аветисян
В статье представлен и оценен новый метод получения начальных приближений для расчётов ядерных электронных орбиталей (NEO-DFT), основанный на аналитическом решении трёхмерного гармонического осциллятора.
Разработанное приближение превосходит существующие методы в ряде случаев и открывает путь к более быстрой сходимости SCF-процедуры в расчётах плотности.
Эффективная сходимость самосогласованных уравнений в многокомпонентных методах среднего поля часто затруднена выбором начальных приближений для квантовых ядер. В работе, озаглавленной ‘Initial Guesses for Multicomponent Mean-Field Methods: Assessment and New Developments’, предпринято систематическое сравнение существующих подходов и разработаны новые приближения, основанные на аналитических решениях трехмерного квантового гармонического осциллятора. Показано, что изотропная версия предложенного начального приближения превосходит существующие методы в расчетах теории функционала плотности ядерно-электронных орбиталей (NEO-DFT), использующих одновременную процедуру самосогласования. Может ли данный подход стать надежным инструментом для повышения скорости сходимости в сложных расчетах NEO-DFT и открыть новые возможности для моделирования ядерных систем?
Начальные Приближения: Вызов Зависимости от Начальной Точки
В основе современных химических расчетов, известных как вычисления электронной структуры, лежит итеративный подход к решению сложных уравнений. Однако, эффективность и надежность этих вычислений напрямую зависят от выбора начальной точки — так называемой «начальной догадки». Итерационные решатели, используемые для определения электронной структуры вещества, подобно исследователям, ищущим оптимальный путь, нуждаются в разумном старте. Неудачная начальная догадка может существенно замедлить процесс поиска решения или привести к получению нефизичного, ошибочного результата. Таким образом, подбор подходящей начальной точки является фундаментальным шагом, определяющим точность и вычислительную стоимость расчетов электронной структуры, и представляет собой критический аспект в современной квантовой химии.
Неудачные начальные приближения в электронных расчетах могут приводить к неспособности алгоритма сойтись к решению, то есть к невозможности получения численного ответа. Но даже если сходимость и достигается, плохо выбранное начальное приближение способно привести к нефизичным результатам, далеким от реального состояния исследуемого объекта. Это особенно критично для сложных молекулярных систем, где энергия поверхности потенциальной энергии имеет множество локальных минимумов. В таких случаях, расчет может застрять в локальном минимуме, не находя истинный минимум, соответствующий стабильному состоянию системы, что существенно снижает точность и эффективность вычислений и требует значительных вычислительных ресурсов для поиска корректного решения.
Качество исходного приближения оказывает непосредственное влияние на вычислительные затраты при проведении электронно-структурных расчетов. Неудачно подобранное начальное состояние может существенно замедлить процесс сходимости итерационных алгоритмов, а в некоторых случаях — и вовсе привести к невозможности получения решения. Это создает критическое узкое место во многих квантово-химических рабочих процессах, поскольку значительная часть времени и ресурсов может быть потрачена не на само решение уравнения, а на поиск подходящего начального приближения. Оптимизация методов генерации исходных состояний, таким образом, является ключевой задачей для повышения эффективности и масштабируемости расчетов, особенно при моделировании сложных молекулярных систем и процессов.

Строительные Блоки: Установленные Методы Начальной Аппроксимации
Метод расширенных Хюккеля (Extended Hückel Method) представляет собой простой полуэмпирический подход к генерации предварительной электронной плотности. Он позволяет быстро получить начальное приближение волновой функции, однако, ввиду упрощающих допущений и отсутствия явного учета электронной корреляции, полученная плотность часто оказывается недостаточной для достижения высокой точности в последующих расчетах, особенно для систем со сложной электронной структурой. Данный метод полезен в качестве отправной точки для более сложных вычислений, но требует осторожной интерпретации результатов и, как правило, не обеспечивает достаточной конвергенции без применения дополнительных методов оптимизации.
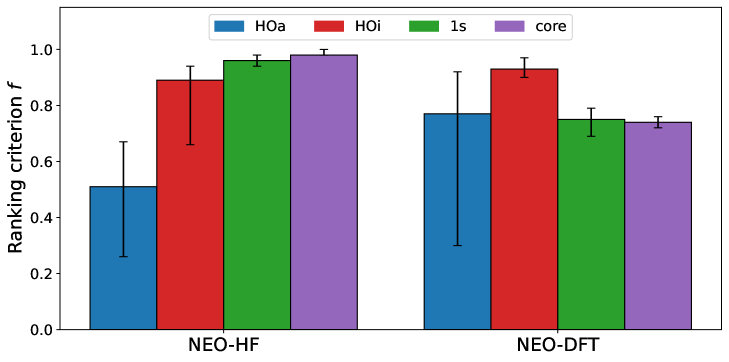
Использование базиса из 1s-подобных функций или плотности, имитирующей электронное ядро, предоставляет минимальные, но физически обоснованные начальные приближения для расчетов. В тестах NEO-HF данный подход демонстрирует высокую результативность, достигая значения ff-rank в 0.98. Однако, следует учитывать, что такая простота может ограничивать применимость метода к более сложным системам, где требуется большая гибкость для адекватного описания электронной структуры и корреляции.
Трёхмерный гармонический осциллятор (ГГО) предоставляет более сложную начальную точку для расчётов, чем методы, использующие простые базисные функции или ядерную плотность. В основе подхода лежит использование чётко определённой поверхности потенциальной энергии, описывающей поведение частиц в трёхмерном пространстве. Это позволяет построить более реалистичное начальное распределение электронов, учитывающее их взаимодействие и энергетические уровни. В отличие от методов, базирующихся на однотипных функциях, ГГО предоставляет возможность гибко моделировать электронную плотность, что особенно важно для систем со сложной электронной структурой и значительным электронным корреляционным вкладом. Начальное распределение, полученное с использованием ГГО, способствует более быстрой сходимости самосогласованных расчётов и повышает точность полученных результатов.

Уточнение Подхода: Использование Гессиана
Вычисление частной матрицы Гессе, состоящей из вторых производных, позволяет получить более точное представление о поверхности потенциальной энергии. Матрица Гессе описывает кривизну этой поверхности, что критически важно для определения оптимальной геометрии молекулы. Использование информации о вторых производных позволяет построить более адекватное приближение к минимуму энергии, тем самым значительно улучшая начальное предположение для итерационных методов оптимизации, таких как NEO-HF и NEO-DFT. В отличие от использования только первых производных (градиента), учет вторых производных позволяет учитывать не только направление, но и скорость изменения энергии, что повышает эффективность алгоритма и снижает количество необходимых итераций для достижения сходимости.
В подходах NEO-HF и NEO-DFT информация о гессиане (матрице вторых производных) используется итеративно для улучшения начального приближения. Этот процесс позволяет ускорить сходимость алгоритма оптимизации и повысить точность вычислений. Итеративное уточнение начального приближения, основанное на гессиане, заключается в последовательном корректировании координат, направленном на минимизацию энергии системы. Эффективность данной методики заключается в более точном определении направления поиска минимума на поверхности потенциальной энергии, что снижает количество необходимых итераций самосогласованного поля (SCF) для достижения заданной точности.
В расчетах NEO-DFT вариации начального приближения, основанные на гармоническом осцилляторе (HO), продемонстрировали высокую эффективность. В частности, HO Изотропное приближение достигло наивысшего значения ff-rank, составив 0.93, что указывает на значительное улучшение качества начального приближения по сравнению с другими протестированными методами. Данный подход также позволил существенно снизить количество итераций самосогласованного поля (SCF) в расчетах, что свидетельствует о более быстрой сходимости и повышении вычислительной эффективности. Полученные результаты подтверждают, что использование HO Изотропного приближения является предпочтительным методом для получения точных и быстро сходящихся результатов в NEO-DFT расчетах.
Базовые Рамки: Плотность и Перекрытие
Матрица перекрывания, получаемая на основе выбранного базисного набора, предоставляет ключевую информацию о взаимодействиях между атомными орбиталями. Эти взаимодействия напрямую влияют на форму и энергию образующихся молекулярных орбиталей, определяя, как электроны распределяются в молекуле. В частности, величина элементов матрицы перекрывания отражает степень взаимного проникновения волновых функций атомных орбиталей, что, в свою очередь, определяет силу связи между атомами и стабильность молекулы. Чем больше перекрытие, тем сильнее взаимодействие и ниже энергия полученной молекулярной орбитали. Понимание этих принципов необходимо для точного моделирования химических систем и предсказания их свойств, поскольку именно матрица перекрывания формирует основу для вычисления электронной структуры и, следовательно, реакционной способности молекул.
Матрица протонной плотности, тесно связанная с матрицей перекрывания атомных орбиталей, определяет вероятностное распределение протонов в молекуле и, следовательно, оказывает фундаментальное влияние на общую электронную структуру. По сути, она описывает, где наиболее вероятно обнаружить протоны, что, в свою очередь, модифицирует потенциальную энергию электронов и, как следствие, влияет на энергию и форму молекулярных орбиталей. Изменение протонной плотности даже на незначительную величину способно приводить к заметным изменениям в реакционной способности и спектральных характеристиках молекулы, подчеркивая её критическую роль в понимании химических процессов. Таким образом, точное определение матрицы протонной плотности является ключевым этапом в квантово-химических расчетах и необходимо для адекватного моделирования химических систем.
В основе всех методов начального приближения в квантово-химических расчетах лежат фундаментальные концепции плотности и перекрывания. Матрица перекрывания, определяющая взаимодействие между атомными орбиталями, и матрица протонной плотности, отражающая распределение вероятности нахождения протонов, совместно формируют каркас, необходимый для построения молекулярных орбиталей и расчета электронной структуры. Понимание этих базовых принципов позволяет не только интерпретировать результаты расчетов, но и целенаправленно совершенствовать алгоритмы начального приближения, повышая их эффективность и точность применительно к широкому спектру химических систем — от простых молекул до сложных биополимеров и материалов. Именно эти концепции обеспечивают надежную основу для последующих итераций оптимизации и позволяют достичь конвергенции к реалистичным решениям уравнения Шрёдингера.
Исследование, представленное в данной работе, демонстрирует, что надежность вычислений не зависит от сложной предварительной настройки. Авторы предлагают новый подход к выбору начального приближения для расчетов NEO-DFT, основанный на аналитическом решении трехмерного гармонического осциллятора. Это подтверждает идею о том, что робастность возникает сама, её нельзя спроектировать. Как заметил Григорий Перельман: «Порядок не нуждается в архитекторе — он возникает из локальных правил». Предложенный метод, улучшая сходимость расчетов, показывает, что структура системы сильнее контроля отдельных агентов, а устойчивость вычислений определяется внутренней логикой самой модели, а не внешним управлением.
Куда Далее?
Представленная работа, хотя и демонстрирует преимущество нового подхода к начальным приближениям в расчетах NEO-DFT, лишь приоткрывает дверь в область, где контроль над сходимостью алгоритмов остается иллюзорным, а влияние локальных правил — определяющим. Поиск оптимальных начальных приближений — это не попытка навязать решение, а скорее, создание условий, в которых самоорганизация системы приведет к стабильному состоянию. Устойчивость и скорость сходимости зависят не от величины «контроля», а от способности алгоритма адаптироваться к локальным особенностям потенциальной поверхности.
Дальнейшие исследования неизбежно должны быть направлены на расширение класса систем, для которых предложенный подход сохраняет свою эффективность. Ограничения, связанные с выбором базисных функций и спецификой решаемых задач, требуют осмысления. Попытки универсализации, вероятно, обречены на неудачу; более продуктивным представляется развитие библиотеки локально-адаптированных начальных приближений, подобно эволюционирующему набору правил, определяющих поведение сложной системы.
В конечном итоге, задача состоит не в том, чтобы «улучшить» алгоритм, а в том, чтобы создать среду, в которой он может самооптимизироваться. Порядок не нуждается в архитекторе — он возникает из локальных взаимодействий. И задача исследователя — не навязывать этот порядок, а лишь помочь ему проявиться.
Оригинал статьи: https://arxiv.org/pdf/2602.11013.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Сила в Модели: Ограничения Оптимизации в Математических Задачах
- Молекулярный интеллект: проверка химического мышления
- Квантовые вычисления для молекул: оптимизация ресурсов
- QR-разложение для экстремальных матриц: новый взгляд на GPU
- Искусственный интеллект и закон: гармония неизбежна
- Самообучающиеся агенты: Новая эра развития искусственного интеллекта
- Искусственный интеллект, который ищет сам: новая стратегия обучения
- Favia: Искусственный интеллект на страже безопасности кода
- Квантовая устойчивость к ошибкам: новый взгляд на исправление вставок и удалений
- Математика и Искусственный Интеллект: Новые Горизонты Открытий
2026-02-13 03:24