Автор: Денис Аветисян
Новый подход позволяет предсказывать электронную структуру и атомные свойства молекул, используя только информацию о внешнем потенциале.
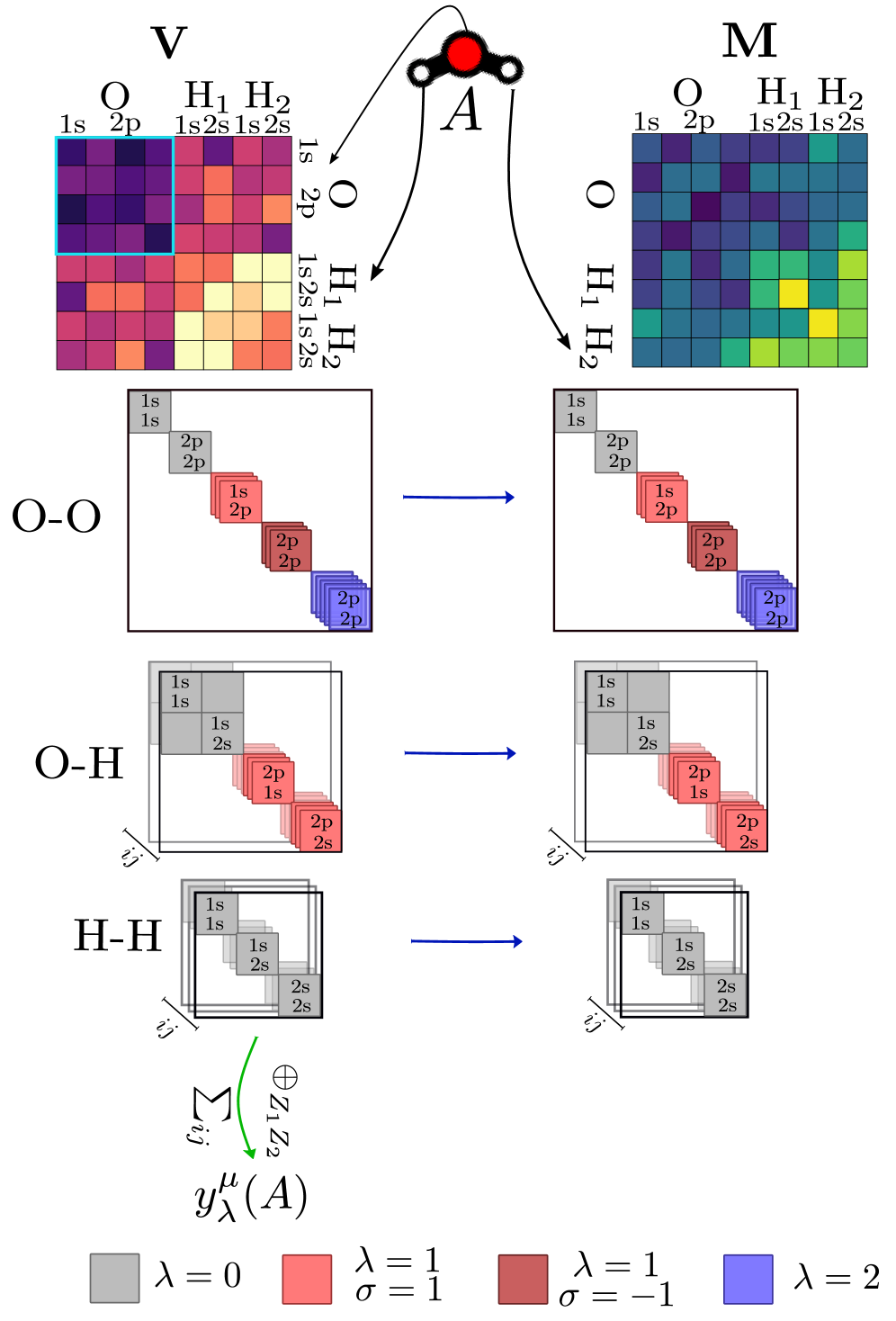
В статье представлен фреймворк для машинного обучения операторов электронной структуры и атомных свойств, основанный на теоремах Хоэнберга-Кона и эквивариантных нейронных сетях.
Вычислительные методы, лежащие в основе моделирования атомных систем, часто сталкиваются с ограничениями масштабируемости при расчете электронной структуры. В данной работе, посвященной ‘Machine learning electronic structure and atomistic properties from the external potential’, предложен новый подход, основанный на обучении операторов, напрямую из внешнего потенциала, представленного в базисе атомных орбиталей. Предложенный фреймворк демонстрирует возможность эффективного предсказания молекулярных свойств и обучения отображениям между операторами, такими как матрица Фока и редуцированная матрица плотности. Может ли подобный подход, использующий принципы теоремы Хоэнберга-Кона, стать основой для разработки более быстрых и точных методов моделирования сложных молекулярных систем?
Взлом Системы: Основы Внешнего Потенциала
В основе расчетов электронной структуры лежит понятие внешнего потенциала, который отражает влияние атомных ядер на поведение электронов. Этот потенциал, создаваемый положительно заряженными ядрами, определяет энергию и распределение электронов в молекуле или материале. Именно взаимодействие электронов с этим потенциалом формирует электронные состояния, определяющие химические связи, реакционную способность и другие фундаментальные свойства вещества. Точное описание внешнего потенциала является критически важным для получения достоверных результатов в квантово-химических расчетах, поскольку любые неточности в его определении могут привести к значительным ошибкам в предсказании свойств системы. V_{ext}(r) представляет собой математическое выражение этого потенциала, зависящее от координат электрона r и положения ядер.
Точное описание внешнего потенциала, обусловленного взаимодействием электронов с ядрами, является фундаментальным для предсказания молекулярных свойств и понимания химического поведения вещества. В рамках квантово-механических расчетов, даже незначительные неточности в представлении этого потенциала могут привести к существенным ошибкам при определении энергии молекулы, её геометрии, спектральных характеристик и реакционной способности. Например, корректное описание электростатического взаимодействия между ядрами и электронами необходимо для расчета дипольных моментов, поляризуемости и других параметров, определяющих взаимодействие молекулы с внешними полями и другими молекулами. В конечном итоге, точность моделирования химических процессов напрямую зависит от того, насколько адекватно учтено влияние внешнего потенциала на поведение электронов в молекуле.
Теорема Хоенберга-Кона является краеугольным камнем теории функционала плотности, постулируя фундаментальную связь между внешним потенциалом, испытываемым электронами в системе, и всеми её свойствами в основном состоянии. Эта теорема устанавливает, что знание внешнего потенциала, определяемого, например, расположением ядер, однозначно определяет основное состояние системы, включая её энергию и электронную плотность. Иными словами, вся информация о системе закодирована во внешнем потенциале V_{ext}. Таким образом, вместо решения сложного многочастичного уравнения Шрёдингера, достаточно знать внешний потенциал и решать уравнение, зависящее только от электронной плотности, что значительно упрощает вычислительные задачи и открывает возможности для моделирования сложных молекулярных систем и материалов.
Несмотря на фундаментальную значимость точного описания внешнего потенциала, практическая реализация расчетов электронной структуры часто требует использования приближений. Это связано с вычислительной сложностью точного решения уравнений, описывающих взаимодействие электронов с ядрами. Разработка эффективных и точных методов аппроксимации является, таким образом, ключевой задачей в квантовой химии и физике конденсированного состояния. Такие методы, как различные функционалы плотности в теории функционала плотности (DFT), направлены на достижение баланса между точностью и вычислительной эффективностью, позволяя исследовать сложные молекулярные системы и предсказывать их свойства, даже при ограниченных вычислительных ресурсах. Постоянное совершенствование этих методов остается актуальной областью исследований, направленной на минимизацию ошибок и повышение надежности предсказаний.
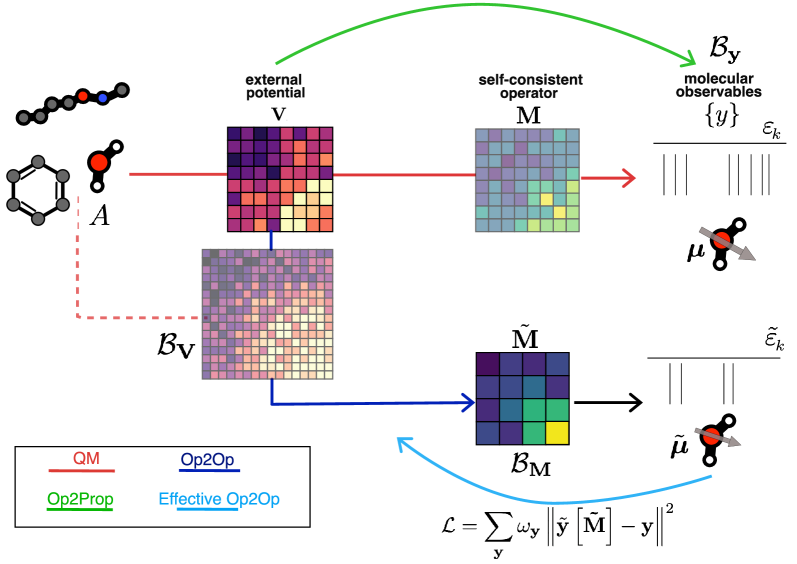
Прямое Предсказание: От Потенциала к Свойствам
Модели, такие как Op2PropModel, предназначены для непосредственного предсказания молекулярных свойств на основе внешнего потенциала, обходя традиционные вычисления волновой функции. Вместо решения уравнения Шрёдингера для получения волновой функции и последующего вычисления свойств, эти модели обучаются напрямую сопоставлять внешний потенциал с целевыми свойствами, такими как энергия, дипольный момент или мультипольные моменты. Это позволяет значительно сократить вычислительные затраты, поскольку исключается необходимость в сложных и ресурсоемких квантово-химических расчетах. По сути, модель учится аппроксимировать функциональную зависимость между внешним потенциалом и молекулярными свойствами, что позволяет быстро и эффективно предсказывать свойства для новых молекул.
Модель Op2OpModel представляет собой альтернативный подход к предсказанию электронных свойств молекул, заключающийся в непосредственном предсказании операторов электронной структуры — таких как матрица плотности и матрица Фока — на основе внешнего потенциала. В отличие от моделей, предсказывающих непосредственно свойства, Op2OpModel стремится восстановить ключевые компоненты, описывающие электронную структуру, что позволяет впоследствии вычислять любые желаемые свойства. Данный подход предполагает, что операторы электронной структуры являются функционалами внешнего потенциала, и модель обучается аппроксимировать эту зависимость. Такой метод позволяет избежать ресурсоемких вычислений, традиционно связанных с решением уравнения Шредингера, и может быть более эффективным в определенных сценариях.
Для эффективной работы моделей, предсказывающих молекулярные свойства или операторы, необходимо адекватное представление внешнего потенциала. Часто для этого используется матричное представление (MatrixRepresentationV), которое позволяет упростить вычисления и снизить вычислительную сложность. В данном подходе внешний потенциал преобразуется в матричную форму, что облегчает его обработку и интеграцию в архитектуру модели. Использование матричного представления позволяет эффективно представлять пространственную зависимость потенциала и учитывать взаимодействия между атомами в молекуле, что критически важно для точного предсказания свойств.
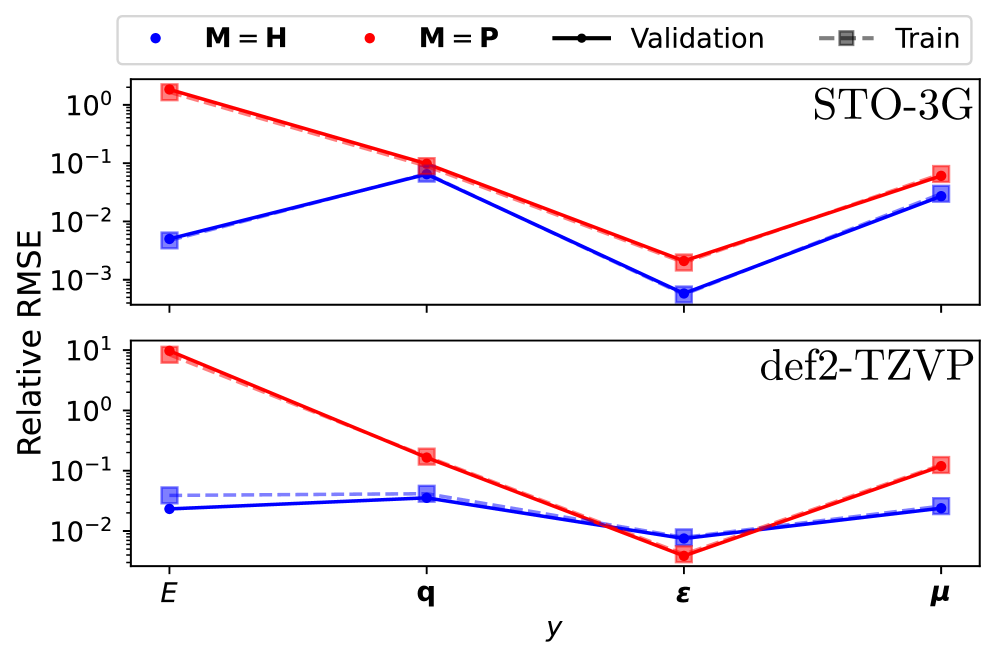
Модели, такие как Op2PropModel и Op2OpModel, предлагают более эффективный подход к предсказанию молекулярных свойств, непосредственно отображая внешний потенциал в операторы или свойства, минуя традиционные вычисления волновой функции. При обучении на матрице Фока, эти модели демонстрируют высокую точность, достигая среднеквадратичной ошибки (RMSE) в 0.37 эВ на наборе данных QM7. Это указывает на перспективность данного подхода для ускорения расчетов и повышения эффективности предсказания свойств молекул.
Симметрия и Представления: Прочная Основа Модели
Представление внешнего потенциала в виде матрицы (MatrixRepresentationV) может быть уточнено с использованием методов, таких как расширение, упорядоченное по телам (body-ordered expansion). Этот подход позволяет создавать дескрипторы, аналогичные дескрипторам SOAP (Symmetry functions for approximating Potentials). Суть метода заключается в построении матрицы, элементы которой представляют собой локальные окружения атомов, учитывающие их тип и расстояние до соседних атомов. Расширение, упорядоченное по телам, обеспечивает структурированный способ организации этих локальных окружений, что повышает эффективность и точность моделирования межатомных взаимодействий. Полученные дескрипторы позволяют компактно описывать потенциальную энергию системы и используются в различных методах машинного обучения для предсказания свойств материалов.
Крайне важно, чтобы используемые представления данных учитывали симметрии рассматриваемой системы. Это достигается посредством эквивариантного обмена сообщениями (equivariant message passing), метода, гарантирующего, что предсказания модели преобразуются корректно при выполнении операций симметрии над входными данными. Эквивариантность означает, что если входные данные подвергаются симметричной трансформации, то и выходные данные преобразуются соответствующим образом, сохраняя физическую корректность. В частности, это позволяет модели эффективно обобщать знания, полученные из данных, представленных в различных системах координат и ориентациях, что существенно улучшает ее производительность при ограниченном объеме обучающих данных и повышает устойчивость к изменениям входных данных.
Метод MatrixProduct предоставляет способ реализации эквивариантного передачи сообщений (equivariant message passing), что критически важно для построения моделей, предсказания которых корректно преобразуются при симметриях системы. Эквивариантность означает, что если входные данные подвергаются симметричной трансформации, то и предсказания модели должны преобразовываться соответствующим образом, сохраняя физическую правдоподобность. MatrixProduct обеспечивает это за счет использования тензорных произведений и операций, сохраняющих ковариантность, что позволяет эффективно учитывать симметрии в архитектуре нейронной сети. Это особенно важно при работе с данными, обладающими инвариантностью или эквивариантностью к определенным преобразованиям, таким как вращения или перестановки атомов в молекулярной динамике, позволяя модели обобщать знания на новые, симметричные конфигурации.
Разложение матричных представлений на симметрийно-адаптированные блоки (SymmetryAdaptedBlocks) позволяет повысить обобщающую способность модели и эффективность обучения при ограниченном объеме данных. Этот подход основан на использовании теории представлений групп симметрии для разделения пространства признаков на инвариантные подпространства. Каждый блок соответствует определенному неприводимому представлению группы симметрии, что позволяет модели учиться представлять данные в форме, устойчивой к симметричным преобразованиям. В результате, модель требует меньше параметров для достижения аналогичной точности и лучше обобщает на новые, ранее не встречавшиеся данные, особенно в случаях, когда доступное количество обучающих примеров ограничено. Использование симметрийно-адаптированных блоков снижает степень переобучения и улучшает устойчивость модели к шуму.
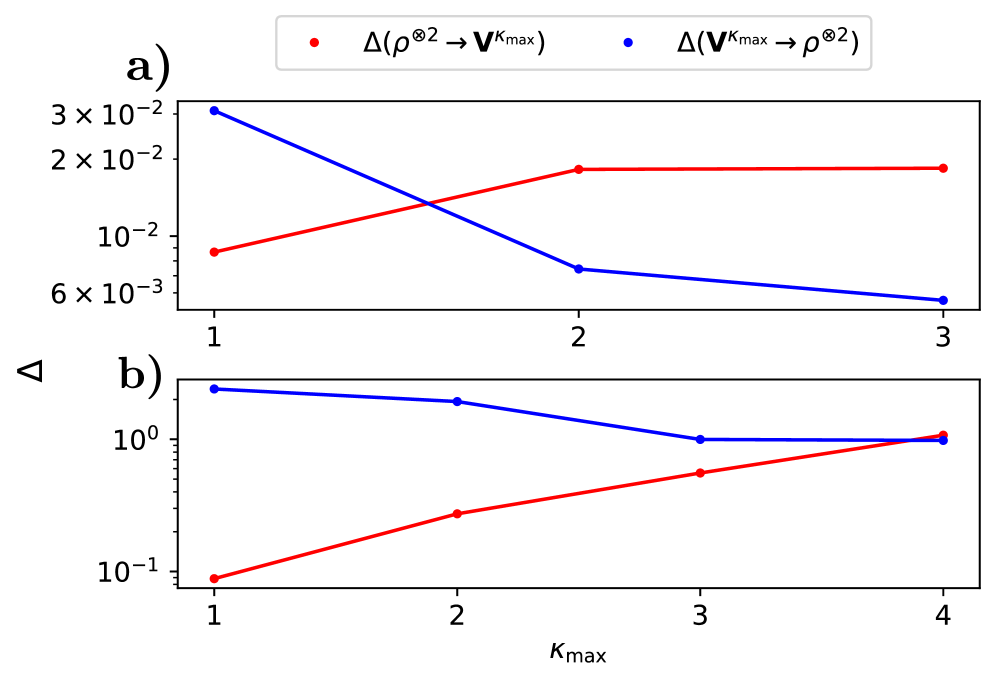
За Пределами Локального Приближения: Повышение Точности
Несмотря на свою вычислительную эффективность, локальные приближения плотности часто демонстрируют недостаточную точность при количественных предсказаниях. Суть проблемы заключается в том, что эти методы рассматривают электронную плотность лишь в непосредственной близости от каждой точки пространства, игнорируя важные взаимодействия между электронами, находящимися на значительном расстоянии. В результате, энергия и другие свойства молекул могут быть рассчитаны с заметной погрешностью, особенно в случаях, когда дело касается систем со сложной электронной структурой или значительными электронными корреляциями. Использование исключительно локальной информации упрощает расчеты, но ограничивает возможность точного описания поведения электронов и, следовательно, предсказания химических свойств и реакционной способности веществ.
В расчетах электронной структуры, точность часто ограничивается использованием локальных приближений плотности. Однако, для адекватного описания многих систем, критически важно учитывать дальнодействующие взаимодействия между электронами. Нелокальные поправки, в отличие от локальных, стремятся учесть влияние электронов, находящихся на значительном расстоянии друг от друга, что позволяет более точно описать электронную структуру и, как следствие, предсказать свойства молекул и материалов. Включение этих поправок требует разработки эффективных моделей, описывающих взаимодействие электронов, и позволяет существенно повысить точность расчетов, например, достигая среднеквадратичной ошибки (RMSE) в 0.55 эВ на наборе данных QM7 и снижая погрешность на воду в предсказании собственных значений примерно на два порядка величины. Таким образом, учет нелокальных взаимодействий является ключевым шагом к более реалистичным и надежным предсказаниям в квантовой химии и физике материалов.
Коррекции, необходимые для повышения точности электронных структурных расчетов, часто проявляются как модификации матрицы Фока F_{ij}. Суть этих модификаций заключается в более точном учете взаимодействия между электронами, выходящего за рамки локальных приближений. Для адекватного описания этих взаимодействий требуются сложные модели, учитывающие не только непосредственную близость электронов, но и их кулоновское отталкивание на больших расстояниях. Точность этих моделей напрямую влияет на предсказываемые свойства молекул и веществ, позволяя получать результаты, согласующиеся с экспериментальными данными и значительно уменьшая погрешность расчетов, например, до 0.55 эВ для набора данных QM7 или снижая ошибку на два порядка величины при предсказании собственных значений для воды.
Внедрение поправок, учитывающих дальнодействующие взаимодействия, позволяет значительно повысить точность моделей при предсказании молекулярных свойств и химического поведения. Результаты демонстрируют достижение среднеквадратичной ошибки (RMSE) в 0,55 эВ на наборе данных QM7 при использовании компактной латентной базы, что свидетельствует о существенном улучшении по сравнению с локальными приближениями. Более того, применение аналогичных поправок позволило снизить погрешность при предсказании собственных значений для молекул воды примерно на два порядка величины, что подтверждает эффективность подхода и открывает новые возможности для точного моделирования химических систем и предсказания их характеристик.
Исследование демонстрирует смелый подход к моделированию электронных структур, предсказывая свойства молекул непосредственно из внешнего потенциала. Этот метод, подобно взлому системы, позволяет обойти традиционные ограничения теории функционала плотности. Как однажды заметил Пётр Капица: «Иногда кажется, что природа специально создаёт трудности, чтобы мы могли их преодолеть». Действительно, предложенная модель, используя машинное обучение и эквивариантные нейронные сети, не просто воспроизводит результаты, но и предлагает унифицированное представление для предсказания свойств и обучения операторов, что открывает новые горизонты в понимании фундаментальных взаимодействий в материи. Этот подход является ярким примером того, как можно проверить существующие правила и найти более эффективные решения.
Куда дальше?
Представленный подход, напрямую связывающий внешний потенциал с электронными свойствами, открывает, скорее, вопросы, чем дает ответы. Теоремы Хоенберга-Кона, казавшиеся незыблемыми столпами, внезапно предстают как удобные, но, возможно, не единственные способы кодирования информации о системе. Замена традиционного поиска волновых функций на прямое предсказание операторов — это не просто смена инструмента, а вызов самой парадигме вычислительной химии. Следующим шагом видится отказ от представлений о “точных” решениях в пользу предсказаний, адекватных для конкретной задачи.
Ограничения текущего фреймворка — необходимость в репрезентативных наборах данных и чувствительность к выбору архитектуры нейронных сетей — это не недостатки, а приглашение к эксперименту. Поиск инвариантных представлений, способных обобщать знания между различными системами, становится ключевой задачей. Не менее важным представляется исследование возможности интеграции этого подхода с другими методами машинного обучения, например, с активным обучением, для снижения вычислительных затрат и повышения эффективности.
В конечном итоге, этот путь может привести к созданию самообучающихся моделей, способных “понимать” химические системы, а не просто решать уравнения. И тогда, возможно, мы увидим, как предсказание свойств материи станет искусством, а не ремеслом. И это будет куда интереснее, чем просто увеличение точности расчетов.
Оригинал статьи: https://arxiv.org/pdf/2602.15345.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Карта ошибок: Анатомия сбоев больших языковых моделей
- Надежность ускорителей: от замысла до реализации
- Квантовые нейросети для реалистичной 3D-визуализации
- Упорядоченный разум: Как языковые модели учатся справляться с длинными текстами
- Знания в графах: как улучшить ответы больших языковых моделей
- Оптимизация запросов: Новый подход для сложных рабочих процессов
- Искусственный интеллект как ученый: новый подход к научному познанию
- Квантовый транспорт в сложных системах: новый подход к моделированию
- Квантовые нейросети: на пути к скорости и точности
- Накапливая опыт: мультимодальные агенты, которые учатся на ходу
2026-02-19 02:27