Автор: Денис Аветисян
Исследователи разработали метод машинного обучения, позволяющий эффективно прогнозировать стабильность сложных наночастиц, используя ограниченные данные расчетов из первых принципов.
Разработанная система использует слой-разрешенные дескрипторы и градиентный бустинг для точной оценки конфигурационной стабильности наночастиц.
Стабильность наночастиц, обусловленная сложным составом и конфигурационным пространством, представляет собой значительную проблему для материаловедения. В работе, озаглавленной ‘Interpretable Machine Learning of Nanoparticle Stability through Topological Layer Embeddings’, предложен новый подход к прогнозированию стабильности наночастиц, основанный на машинном обучении с использованием слоевого дескриптора и градиентного бустинга. Данный фреймворк позволяет эффективно идентифицировать наиболее стабильные конфигурации, используя лишь ограниченное количество расчетов на основе теории функционала плотности. Какие перспективы открываются для активного обучения и исследования конфигурационного пространства сложных наночастиц с помощью предложенного интерпретируемого подхода?
Постановка задачи: предсказание стабильности сплавов
Предсказание стабильности металлических сплавов, таких как Al70Co10Fe5Ni10Cu5, имеет первостепенное значение для разработки новых материалов с заданными свойствами. Однако, традиционные методы, основанные на расчетах термодинамических свойств и моделировании равновесных состояний, требуют значительных вычислительных ресурсов. Это связано с необходимостью учета множества возможных конфигураций атомов в сплаве и сложными взаимодействиями между ними. Поиск наиболее стабильной структуры в пространстве всех возможных вариантов становится непомерно затратным по времени и вычислительной мощности, особенно для сплавов сложного состава и высокой энтропии. Таким образом, разработка эффективных и экономичных методов предсказания стабильности сплавов остается актуальной задачей материаловедения и физики твердого тела.
Стабильность металлических сплавов, таких как Al70Co10Fe5Ni10Cu5, определяется сложным взаимодействием нескольких факторов. Химический беспорядок, возникающий из-за случайного распределения атомов различных элементов, существенно влияет на энергию системы. Поверхностная сегрегация, когда определенные атомы стремятся к поверхности сплава, изменяет локальные химические составы и, следовательно, стабильность. Наконец, координационная топология, описывающая, как атомы связаны с ближайшими соседями, формирует структуру сплава и определяет его устойчивость к деформациям и фазовым превращениям. Сочетание этих факторов создает многомерный “ландшафт” стабильности, где даже небольшие изменения в составе или структуре могут привести к значительным изменениям в свойствах материала, что делает предсказание стабильности сплава сложной задачей.
Точное прогнозирование стабильности сплавов сопряжено с необходимостью исследования колоссального конфигурационного пространства, что делает полный перебор всех возможных состояний практически невозможным. Представьте себе, что каждый атом в сплаве может занимать множество различных позиций и взаимодействовать с окружающими его атомами — количество комбинаций быстро становится астрономическим. Даже при использовании самых мощных вычислительных ресурсов, полный перебор всех конфигураций требует неприемлемо большого времени и ресурсов. Поэтому исследователи вынуждены разрабатывать инновационные методы, такие как методы Монте-Карло и молекулярной динамики, а также применять статистические подходы для эффективного поиска наиболее стабильных конфигураций, фокусируясь на наиболее вероятных состояниях и используя приближенные модели для снижения вычислительной сложности.
Переосмысление задачи: ранжирование конфигураций
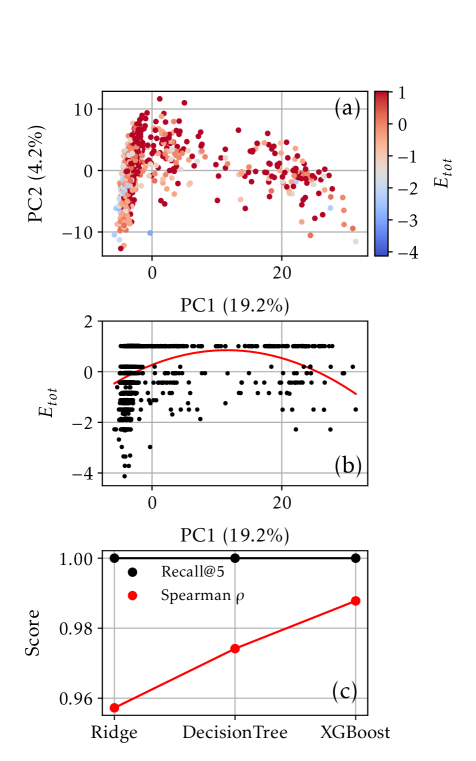
Вместо прямого предсказания абсолютных энергий, мы формулируем задачу как ранжирование конфигураций наночастиц по их относительной стабильности. Такой подход позволяет избежать проблем, связанных с накоплением ошибок при предсказании абсолютных значений, и фокусируется на определении наиболее и наименее стабильных структур в заданном наборе. Вместо того, чтобы предсказывать значение энергии для каждой конфигурации, алгоритм определяет порядок конфигураций от наиболее стабильной к наименее стабильной. Это позволяет использовать алгоритмы машинного обучения, оптимизированные для задач ранжирования, что потенциально повышает точность и эффективность модели по сравнению с задачами регрессии, где необходимо предсказывать непрерывные значения.
Вместо прогнозирования абсолютных энергий, сформулировав задачу как ранжирование конфигураций наночастиц по относительной стабильности, становится возможным использовать модели машинного обучения, оптимизированные для задач упорядочивания (ranking). Традиционные регрессионные модели, предназначенные для предсказания непрерывных значений, менее эффективны в данной ситуации, поскольку ключевым является определение порядка стабильности между различными конфигурациями, а не точное значение энергии каждой из них. Алгоритмы ранжирования напрямую оптимизируются для корректного упорядочивания объектов, что позволяет добиться более высокой точности и эффективности при решении задачи оценки стабильности наночастиц. Это особенно актуально, когда количество возможных конфигураций велико, и требуется быстро определить наиболее стабильные из них.
В качестве эталонных данных для начального обучения и валидации подхода ранжирования используются расчеты в рамках теории функционала плотности (DFT). DFT позволяет получить точные значения энергии различных конфигураций наночастиц, которые служат основой для определения их относительной стабильности. Эти значения энергии используются для создания обучающего набора данных, необходимого для тренировки моделей машинного обучения, предназначенных для задачи ранжирования. Валидация точности моделей производится путем сравнения предсказанного ранжирования с ранжированием, полученным на основе результатов DFT-расчетов. Использование DFT гарантирует, что модели машинного обучения обучаются на физически обоснованных данных, что повышает надежность и точность предсказаний.
Детали реализации: слой-разрешающие дескрипторы и машинное обучение
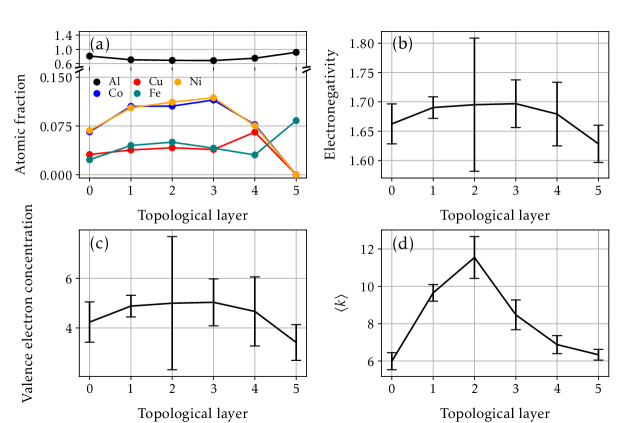
Дескриптор, разрешающий по слоям, представляет собой метод анализа наночастиц, при котором структура разделяется на последовательные топологические слои, определяемые на основе расположения атомов и их координационного окружения. Такой подход позволяет модели учитывать пространственные вариации в атомной среде, что особенно важно для описания поверхностных атомов и краевых эффектов. В отличие от глобальных дескрипторов, которые характеризуют всю наночастицу одним вектором, данный метод предоставляет детальную информацию о локальном окружении каждого атома в зависимости от его положения в слоях наночастицы. Это обеспечивает более точное описание энергетических свойств и стабильности различных конфигураций, поскольку учитывает различия в координационном числе, межатомных расстояниях и углах связи в разных слоях.
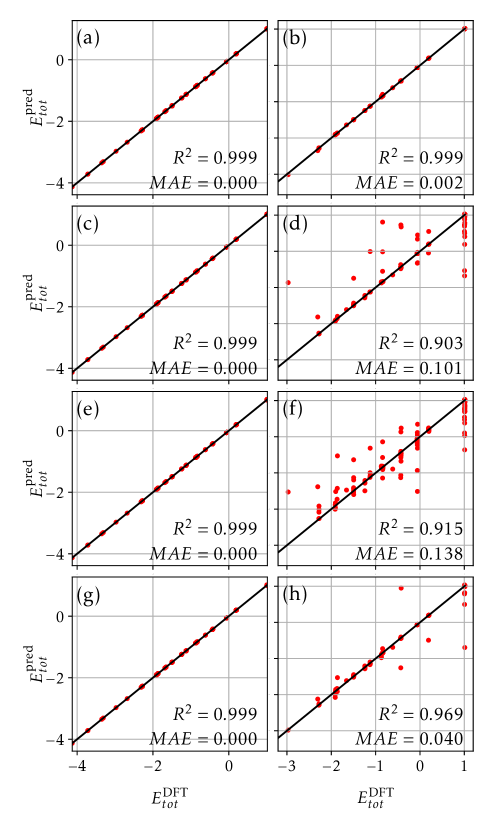
В качестве входных данных для модели XGBoost используется дескриптор, разрешающий слои наночастиц. XGBoost представляет собой алгоритм градиентного бустинга на деревьях решений, который обучается предсказывать относительную стабильность различных конфигураций. В процессе обучения модель анализирует значения дескриптора для каждой конфигурации и соотносит их с энергией, рассчитанной методом DFT, устанавливая связь между структурой и стабильностью. Использование ансамбля деревьев решений позволяет модели эффективно обрабатывать сложные нелинейные зависимости и обеспечивать высокую точность прогнозирования энергии, что необходимо для оценки стабильности наночастиц.
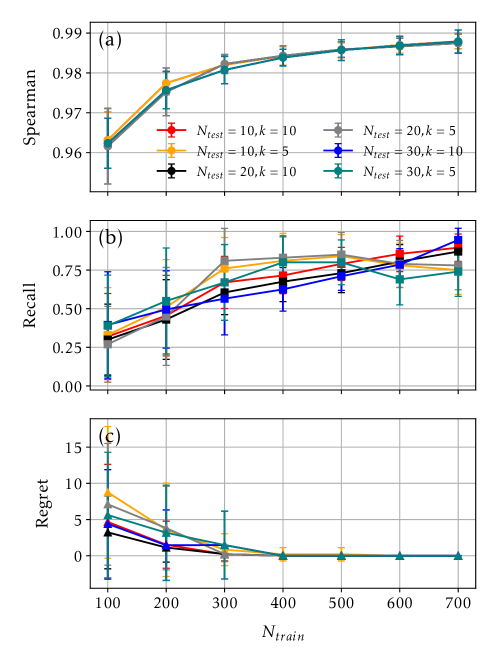
Для повышения точности модели и снижения вычислительных затрат реализован метод активного обучения. Данный подход позволяет интеллектуально отбирать конфигурации для расчетов с использованием теории функционала плотности (DFT). Модель, обученная на относительно небольшом количестве структур DFT (200-300), демонстрирует высокий коэффициент корреляции Спирмена ≥ 0.97, что указывает на ее способность к точной оценке относительной стабильности конфигураций даже при ограниченном объеме обучающих данных. Это достигается за счет итеративного процесса, в котором модель предсказывает стабильность, а наиболее информативные конфигурации выбираются для проведения DFT-расчетов и последующей переобучения модели.
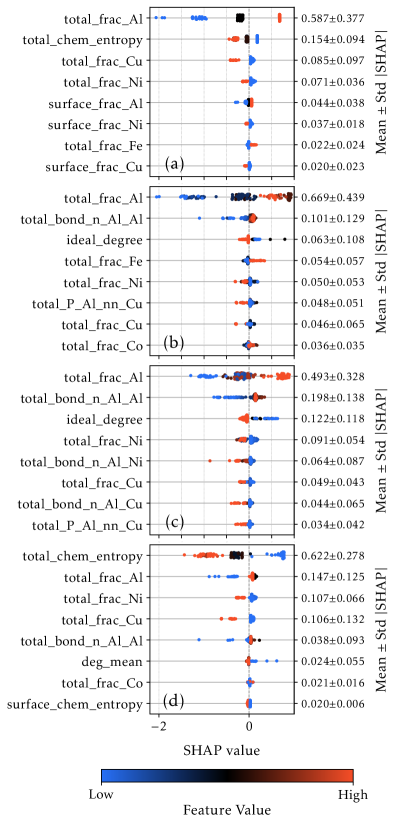
Интерпретация предсказаний: анализ SHAP
Для интерпретации предсказаний модели XGBoost был применен анализ SHAP, позволивший выявить ключевые факторы, определяющие стабильность сплавов. Этот метод позволяет оценить вклад каждого признака в конечное предсказание, что даёт возможность понять, какие структурные характеристики наиболее сильно влияют на стабильность. Анализ SHAP не просто показывает, какие признаки важны, но и указывает направление их влияния — то есть, как увеличение или уменьшение значения конкретного признака сказывается на предсказанной стабильности сплава. Это позволяет исследователям не только предсказывать стабильность новых материалов, но и целенаправленно изменять их состав и структуру для достижения желаемых свойств, раскрывая взаимосвязи между химическим составом, кристаллической структурой и термодинамической устойчивостью.
Количественная оценка вклада каждой характеристики позволяет выявить ключевые структурные особенности, определяющие стабильность сплавов. Анализ показывает, что такие параметры, как энергия связи, атомные радиусы и электроотрицательность компонентов, оказывают наибольшее влияние на предсказываемую стабильность кристаллических решеток. Выявление этих доминирующих факторов не только углубляет понимание фундаментальных механизмов, управляющих стабильностью сплавов, но и предоставляет ценные сведения для рационального дизайна новых материалов с заданными свойствами. Например, моделирование демонстрирует, что определенные комбинации элементов, характеризующиеся сбалансированными значениями указанных параметров, приводят к значительно более стабильным структурам, что может быть использовано для создания высокоэффективных и долговечных сплавов.
Оценка производительности модели, основанная на корреляции рангов Спирмена, подтвердила её способность точно ранжировать различные конфигурации сплавов. Примечательно, что модель демонстрирует почти абсолютный показатель Top-5 Recall, что означает, что наиболее стабильные конфигурации практически всегда попадают в топ-5 предсказаний. Более того, анализ показал крайне низкий уровень сожаления (regret), указывающий на минимальную разницу между предсказанными и фактическими значениями стабильности. Впечатляющим является тот факт, что модель достигает таких высоких показателей, используя для обучения лишь 200-300 структур, что подчеркивает её эффективность и потенциал для быстрого скрининга новых материалов.
Ускорение материаловедческих открытий: скрининг Top-k
Обученная модель позволяет проводить так называемый Top-k скрининг, что представляет собой эффективный метод быстрой идентификации наиболее перспективных кандидатов на роль наночастиц для дальнейшего изучения. Этот подход заключается в предварительной оценке большого числа потенциальных материалов с использованием алгоритмов машинного обучения, после чего отбираются лишь k наиболее многообещающих вариантов для более детального анализа. Такой способ значительно сокращает время и вычислительные затраты, связанные с традиционными методами материаловедения, позволяя исследователям сосредоточиться на наиболее перспективных направлениях и ускорить процесс открытия новых сплавов с заданными свойствами. Данный метод позволяет не только сократить время поиска, но и расширить область исследуемых материалов, открывая возможности для создания инновационных решений в различных областях науки и техники.
Традиционный дизайн новых сплавов требует огромных вычислительных ресурсов, поскольку необходимо исследовать бесчисленное количество комбинаций элементов и структур. Однако, применяемый подход значительно снижает эту вычислительную нагрузку, позволяя сосредоточиться на наиболее перспективных кандидатах. Благодаря эффективному отбору, количество необходимых расчетов сокращается в разы, что ускоряет процесс открытия материалов с заданными свойствами. Это особенно важно при исследовании сложных многокомпонентных сплавов, где перебор всех возможных вариантов практически невозможен. Уменьшение вычислительных затрат не только экономит время и энергию, но и открывает доступ к исследованию более широкого спектра материалов, стимулируя инновации в области материаловедения.
Сочетание машинного обучения с расчетами из первых принципов открывает принципиально новый подход к разработке материалов. Традиционно, поиск новых сплавов требовал огромных вычислительных ресурсов и времени, поскольку необходимо было моделировать свойства бесчисленных комбинаций элементов. Однако, используя алгоритмы машинного обучения для предварительного отбора наиболее перспективных кандидатов, а затем уточняя их свойства с помощью высокоточных расчетов на основе квантовой механики, можно значительно сократить объем необходимых вычислений. Этот симбиоз позволяет не просто случайным образом перебирать варианты, а целенаправленно исследовать области химического пространства, где наиболее вероятно обнаружение материалов с заданными характеристиками, что в конечном итоге ускоряет процесс создания инновационных сплавов и материалов будущего.
Исследование демонстрирует, что понимание стабильности наночастиц требует целостного подхода, а не фокусировки на отдельных аспектах. Подобно тому, как структура определяет поведение системы, предложенный фреймворк машинного обучения, использующий слой-разрешенные дескрипторы, позволяет оценить конфигурационную стабильность наночастиц с ограниченным количеством расчетов на основе теории функционала плотности. Как заметил Альберт Эйнштейн: «Самое прекрасное, что мы можем испытать — это тайна. Истинную науку можно определить как стремление к этой тайне». Стремление к пониманию стабильности наночастиц, таким образом, является поиском порядка в кажущемся хаосе, что отражает суть научного исследования.
Куда двигаться дальше?
Представленный подход, фокусируясь на слоистых дескрипторах и градиентном бустинге, демонстрирует эффективность в предсказании стабильности наночастиц при ограниченном количестве расчетов из первых принципов. Однако, элегантность этой простоты обнажает и лежащие в тени ограничения. Каждая новая зависимость от конкретного дескриптора — это скрытая цена свободы от вычислительных затрат. Очевидно, что универсальность подобного подхода нуждается в дальнейшей проверке на более сложных, многокомпонентных системах, где взаимодействие между слоями становится определяющим фактором.
Будущие исследования должны быть направлены на расширение пространства дескрипторов, включая не только геометрические параметры, но и информацию об электронной структуре, а также на разработку методов активного обучения, которые позволят целенаправленно выбирать наиболее информативные расчеты. Важно помнить, что стабильность — это не статичное свойство, а динамический процесс, и учет влияния температуры и внешних полей может значительно повысить точность предсказаний.
В конечном итоге, структура данных определяет поведение модели. Поиск оптимального баланса между информативностью дескрипторов, вычислительной сложностью и обобщающей способностью — это не просто техническая задача, а философский поиск гармонии между точностью и эффективностью. Иначе говоря, задача не в том, чтобы построить сложную систему, а в том, чтобы понять, как работает простая.
Оригинал статьи: https://arxiv.org/pdf/2602.17528.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Эхо чёрных дыр: как квантовая гравитация меняет гравитационные волны
- Трансформация нейросетей: от плотных моделей к разреженным экспертам без обучения
- Сияние фотонов: новый уровень точности в предсказаниях столкновений частиц
- Биосети в руках ИИ: Автоматизация системной фармакологии
- Укрощение Бесконечности: Алгебраические Инструменты для Кватернионов и За их Пределами
- Квантовая Физика и Безопасность: Анализ Последних Новостей
- Оптические Тензорные Ядра: Путь к Масштабируемым Вычислениям
- Квантовые ограничения в хаотичных сплавах: взгляд на Si/SiGe/Si
- Шёпот языков: как дрессировать цифрового голема для забытых наречий.
- Квантовое моделирование: от проблемных решений к универсальному программному обеспечению
2026-02-22 04:11