Автор: Денис Аветисян
Исследователи представили аппаратную реализацию полуэмпирических методов электронной структуры на FPGA, значительно повысив скорость вычислений.
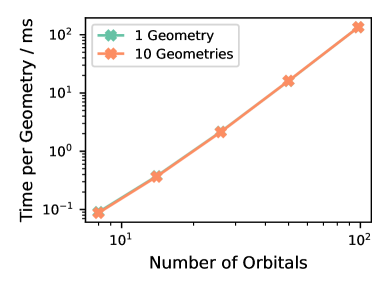
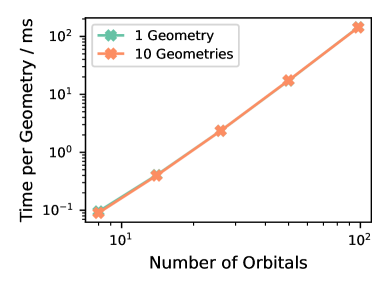
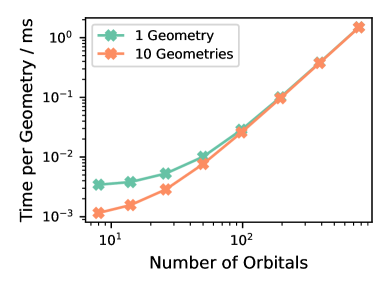
Представлена аппаратная реализация метода, ускоряющая построение гамильтониана, но требующая оптимизации алгоритмов для диагонализации плотных матриц.
Вычислительные затраты, связанные с квантово-химическими расчетами, становятся все более серьезным препятствием для моделирования сложных молекулярных систем и материалов. В данной работе, посвященной ‘A Hardware-Native Realisation of Semi-Empirical Electronic Structure Theory on Field-Programmable Gate Arrays’, представлено первое аппаратное воплощение полуэмпирической теории электронной структуры на базе программируемой пользователем вентильной матрицы (FPGA). Разработанная архитектура, реализующая теорию Extended Hückel и DFTB0, обеспечивает существенное ускорение генерации гамильтониана по сравнению с современными процессорами. Не решив полностью проблему эффективной диагонализации плотных матриц, данное исследование открывает новые перспективы для создания энергоэффективных и масштабируемых аппаратных решений в области квантохимического моделирования?
Вычислительные Пределы: Узкое Место Электронной Структуры
Высокоточные методы ab initio, несмотря на свою мощь в предсказании свойств материалов и молекул, сталкиваются с серьезными ограничениями, обусловленными вычислительной сложностью расчета интегралов электронного отталкивания. Эти интегралы, описывающие взаимодействие между электронами в системе, требуют огромных ресурсов для своего точного вычисления, особенно при моделировании систем, содержащих большое количество электронов. Сложность возрастает пропорционально N^4, где N — число базисных функций, что делает применение этих методов крайне затратным по времени и памяти даже для умеренно сложных молекул и материалов. В результате, возможности моделирования ограничиваются размером системы и длительностью расчетов, что препятствует изучению сложных химических процессов и поиску новых материалов с заданными свойствами. Таким образом, вычислительные затраты, связанные с интегралами электронного отталкивания, представляют собой узкое место в современных расчетах электронной структуры.
Вычислительные затраты при моделировании электронного строения материалов в значительной степени обусловлены ограничениями пропускной способности памяти и недостаточной арифметической интенсивностью вычислений. Суть проблемы заключается в том, что для расчета взаимодействия электронов необходимо обрабатывать огромные объемы данных, что требует постоянного обмена информацией между процессором и памятью. При этом, операции с этими данными, несмотря на их сложность, не всегда требуют большого количества арифметических операций на единицу переданной информации. В результате, узким местом становится не скорость процессора, а скорость, с которой данные могут быть получены и обработаны, что существенно ограничивает возможность моделирования сложных систем, таких как большие молекулы или материалы с дефектами, и замедляет прогресс в материаловедении и химии.
Традиционные методы расчета электронной структуры сталкиваются с серьезными трудностями при одновременном обеспечении высокой точности и скорости, необходимой для высокопроизводительного скрининга и молекулярной динамики. Повышение точности обычно требует экспоненциального увеличения вычислительных ресурсов, что делает невозможным моделирование больших систем или проведение длительных временных симуляций. В то время как упрощенные подходы позволяют обрабатывать большее количество молекул или увеличить продолжительность моделирования, они часто приводят к неприемлемым потерям точности, что снижает надежность полученных результатов. В связи с этим, возникает потребность в разработке новых алгоритмов и вычислительных стратегий, способных эффективно балансировать между точностью и вычислительной стоимостью, открывая возможности для ускоренного открытия материалов и углубленного понимания сложных химических процессов.
Ограничения, накладываемые вычислительными ресурсами при моделировании электронного строения, существенно замедляют прогресс в области материаловедения и химии. Невозможность проводить точные расчеты для сложных систем, включающих большое количество атомов или требующих длительного моделирования динамических процессов, препятствует быстрому открытию новых материалов с заданными свойствами. Например, поиск эффективных катализаторов или разработка новых аккумуляторов требует понимания реакционных механизмов на атомном уровне, что становится крайне сложной задачей при существующих ограничениях. В результате, прогресс в этих областях происходит значительно медленнее, чем мог бы, если бы вычислительные барьеры были преодолены. Поиск и разработка новых материалов и понимание сложных химических процессов, таким образом, напрямую зависят от развития более эффективных вычислительных методов.
Полуэмпирические Методы: Прагматичный Подход к Моделированию
Полуэмпирические методы представляют собой прагматичное решение в квантово-химических расчетах, основанное на параметризации гамильтониана. Вместо вычисления всех интегралов «с нуля», эти методы используют заранее вычисленные параметры, полученные из экспериментальных данных или более точных, но вычислительно затратных расчетов. Такой подход существенно снижает вычислительную сложность, позволяя проводить расчеты для систем большего размера, однако это достигается за счет некоторой потери точности. Параметризация гамильтониана позволяет аппроксимировать электронную структуру системы, фокусируясь на наиболее важных взаимодействиях и пренебрегая менее значимыми, что является компромиссом между точностью и скоростью вычислений.
Полуэмпирические методы, такие как Density Functional Tight Binding (DFTB) и Extended Hückel Theory (EHT), существенно снижают вычислительные затраты за счет использования предварительно вычисленных интегралов и различных приближений. Вместо решения уравнения Шредингера “с нуля”, эти методы оперируют упрощенными выражениями, где многие параметры, определяющие взаимодействие между атомами, берутся из экспериментальных данных или результатов более сложных расчетов. Например, в DFTB, матричные элементы, описывающие взаимодействие, вычисляются на основе заранее определенных функций и параметров, а EHT использует упрощенную формулу для вычисления энергии \epsilon = \alpha + \sum_{i \neq j} p_{ij}, где α — энергия атома, а p_{ij} — интеграл перекрытия атомных орбиталей. Такой подход позволяет проводить расчеты для систем, содержащих сотни или даже тысячи атомов, что недоступно для методов, основанных на полном решении уравнения Шредингера.
Построение гамильтониана, являясь ключевым этапом в полуэмпирических методах, значительно упрощается за счет использования параметризации и предварительно вычисленных интегралов. Это позволяет существенно сократить вычислительные затраты по сравнению с ab initio подходами. Вместо вычисления всех элементов матрицы гамильтониана напрямую, полуэмпирические методы используют параметры, такие как параметры Слейтера-Костера, для определения взаимодействий между атомами. Такой подход позволяет избежать трудоемких расчетов, сохраняя при этом приемлемую точность для многих задач, особенно при моделировании больших систем, где скорость вычислений является критическим фактором. H_{ij} = a_{ij} + b_{ij}r_{ij} + c_{ij}r_{ij}^2 — типичное представление упрощенного элемента матрицы гамильтониана.
Параметры Слейтера-Костера являются ключевыми элементами в полуэмпирических методах, определяя силу и характер взаимодействия между атомными орбиталями. Эти параметры, как правило, представляют собой матричные элементы H_{\mu\nu}, описывающие перекрывание и потенциальную энергию между орбиталями μ и ν. Значения параметров Слейтера-Костера зависят от типов атомов и расстояния между ними, и часто определяются путем подгонки к экспериментальным данным или более точным квантово-механическим расчетам. Использование этих параметров позволяет упростить вычисление гамильтониана системы, существенно снижая вычислительные затраты при моделировании электронных структур материалов.

Аппаратное Ускорение: От Графических Процессоров к FPGA
Графические процессоры (GPU) стали стандартом для ускорения расчетов в электронной структуре благодаря своим возможностям параллельной обработки. В отличие от центральных процессоров (CPU), которые оптимизированы для последовательного выполнения задач, GPU содержат тысячи небольших ядер, способных одновременно выполнять множество операций. Это делает их особенно эффективными для задач, таких как решение систем линейных уравнений и диагонализация матриц, которые являются ключевыми этапами в расчетах электронной структуры. Параллельная архитектура GPU позволяет значительно сократить время вычислений по сравнению с CPU, особенно для больших систем и сложных моделей. O(n^3) сложность многих алгоритмов электронной структуры эффективно снижается за счет использования GPU.
Полевые программируемые вентильные матрицы (FPGA) представляют собой альтернативный подход к аппаратному ускорению вычислений, позволяющий реализовать вычисления непосредственно на аппаратном уровне с использованием специализированных микроархитектур. В отличие от графических процессоров (GPU), которые оптимизированы для параллельной обработки графики, FPGA позволяют спроектировать логическую схему, адаптированную непосредственно под конкретную задачу. Это достигается путем программирования конфигурации внутренних логических элементов и соединений FPGA, что позволяет создавать пользовательские вычислительные блоки и оптимизировать поток данных для достижения максимальной производительности. Такой подход позволяет избежать накладных расходов, связанных с универсальной архитектурой GPU, и добиться более высокой энергоэффективности и скорости выполнения специализированных алгоритмов.
Реализация вычислений непосредственно на структуре FPGA, поддерживаемая потоковой архитектурой, обеспечивает максимальную производительность. Данная работа демонстрирует первую аппаратную реализацию полуэмпирического метода электронной структуры на FPGA, что позволило достичь пиковой пропускной способности при генерации гамильтониана. Потоковая архитектура позволяет обрабатывать данные последовательно, минимизируя задержки и эффективно используя ресурсы FPGA. Это отличается от традиционных подходов, использующих CPU или GPU для выполнения тех же вычислений, где данные должны перемещаться между памятью и процессором. Достигнутая пиковая пропускная способность свидетельствует о потенциале FPGA для значительного ускорения вычислений в квантовой химии и материаловедении.
В основе ускорения расчетов в электронных структурах часто лежат эффективные алгоритмы диагонализации, такие как метод Якоби. Данный метод, итеративно приводящий симметричную матрицу к диагональному виду посредством ортогональных преобразований, является ключевым этапом в решении задачи собственных значений Hv = \lambda v, где H — матрица Гамильтона, v — собственный вектор, а λ — собственное значение. Эффективность метода Якоби напрямую влияет на скорость вычисления энергетических уровней и волновых функций, что делает оптимизацию его реализации критически важной для аппаратного ускорения расчетов.
Влияние на Дизайн и Открытие Материалов
Ускорение расчетов электронной структуры посредством использования FPGA и GPU открывает принципиально новые возможности для высокопроизводительного скрининга обширных химических пространств. Традиционные методы часто сталкиваются с вычислительными ограничениями при исследовании огромного количества потенциальных материалов, однако применение специализированного аппаратного обеспечения позволяет значительно увеличить скорость моделирования. Это, в свою очередь, позволяет исследователям исследовать гораздо большее количество химических соединений и кристаллических структур, идентифицируя вещества с заданными свойствами, такими как высокая электропроводность, механическая прочность или каталитическая активность. Такой подход не только сокращает время, необходимое для открытия новых материалов, но и снижает затраты на экспериментальные исследования, направляя ресурсы на наиболее перспективные кандидаты, отобранные в ходе виртуального скрининга.
Ускорение вычислительных процессов открывает принципиально новые возможности для создания материалов с заданными характеристиками. Благодаря значительному повышению скорости скрининга огромного количества химических соединений, исследователи получают возможность быстро идентифицировать перспективные кандидаты для различных применений — от высокоэффективных аккумуляторов до сверхпроводящих материалов. Этот подход позволяет сократить время, необходимое для разработки и тестирования новых материалов, что особенно важно в условиях постоянно растущих технологических требований. Возможность быстрого моделирования и анализа свойств веществ способствует целенаправленному проектированию материалов, оптимизированных под конкретные задачи, и существенно расширяет границы инноваций в материаловедении.
Эффективное моделирование играет ключевую роль в постижении сложных химических процессов и оптимизации характеристик материалов. Понимание взаимодействия атомов и молекул на фундаментальном уровне требует значительных вычислительных ресурсов, однако, благодаря развитию алгоритмов и аппаратного обеспечения, стало возможным проводить детальный анализ даже самых сложных систем. Точные симуляции позволяют предсказывать свойства материалов, такие как прочность, электропроводность и термостойкость, до их фактического синтеза, существенно сокращая время и затраты на разработку новых технологий. Возможность моделировать динамическое поведение материалов в различных условиях позволяет оптимизировать их состав и структуру для достижения заданных характеристик, открывая новые горизонты в материаловедении и инженерии.
Сочетание передовых вычислительных методов с искусственным интеллектом в виде силовых полей открывает новые возможности для моделирования динамики молекул с беспрецедентной точностью и эффективностью. Данный подход позволяет значительно ускорить процессы молекулярной динамики, что особенно важно при изучении сложных систем. Недавние исследования продемонстрировали масштабируемость этой технологии, успешно применив ее к моделированию крупной молекулы C128H258. Это подтверждает возможность использования подобных методов для изучения и прогнозирования поведения сложных материалов, что, в свою очередь, способствует разработке новых материалов с заданными свойствами и оптимизации существующих.
Исследование, представленное в данной работе, демонстрирует возможности аппаратной реализации методов расчета электронной структуры, в частности, с использованием FPGA. Такой подход позволяет значительно ускорить построение гамильтониана, что критически важно для высокопроизводительных вычислений. В контексте этого, уместно вспомнить слова Игоря Евгеньевича Тамма: «В науке важно не только увидеть факт, но и понять его место в системе знаний». Подобно тому, как аппаратная реализация ускоряет отдельные этапы вычислений, понимание взаимосвязи между алгоритмами, аппаратным обеспечением и фундаментальными принципами физики позволяет создавать более эффективные и мощные инструменты для исследования материального мира. Особое внимание в работе уделяется необходимости оптимизации методов плотной диагонализации, что подчеркивает важность комплексного подхода к решению сложных научных задач.
Что дальше?
Представленная работа, демонстрируя аппаратную реализацию полуэмпирических методов электронной структуры на FPGA, обнажает нетривиальную закономерность: ускорение построения гамильтониана оказывается значительно проще, чем эффективная диагонализация плотных матриц. Каждое изображение данных, полученных в ходе экспериментов, скрывает структурные зависимости, требующие глубокого анализа. Ускорение одного этапа лишь подчеркивает узкие места в других, подобно тому, как увеличение разрешения микроскопа выявляет новые детали, требующие интерпретации.
Будущие исследования неизбежно столкнутся с необходимостью разработки специализированных аппаратных средств для решения задач диагонализации. Простое увеличение вычислительных ресурсов не является решением, поскольку фундаментальные ограничения алгоритмов остаются. Вместо этого, представляется перспективным поиск альтернативных подходов, возможно, основанных на итерационных методах или приближенных алгоритмах, адаптированных для FPGA-архитектуры. Интерпретация моделей важнее красивых результатов; ключевым представляется не просто достижение высокой скорости, а понимание границ применимости и точности полученных решений.
Таким образом, дальнейшее развитие данного направления требует не только инженерных усилий по оптимизации аппаратного обеспечения, но и фундаментальных исследований в области численных методов и алгоритмов. Каждая новая реализация на FPGA — это не просто технологический шаг вперед, а возможность глубже понять закономерности, лежащие в основе квантово-механических расчетов.
Оригинал статьи: https://arxiv.org/pdf/2602.11702.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Конфиденциальный анализ больших данных: новый подход к быстрым ответам
- Квантовая физика: От хаоса к пониманию
- Искусственный глаз: Как отличить реальное изображение от сгенерированного ИИ
- Когда большая языковая модель молчит: как избежать галлюцинаций при ответе на вопросы?
- Юридический интеллект на турецком: Новые модели для понимания права
- Разреженность и масштаб: семейство языковых моделей Trinity
- Голос с Акцентом: Управление произношением без акцентированных данных
- Квантовая магия: Революция нулевого уровня!
- Раскрывая потенциал языковых моделей: новый взгляд на оценку
- Преображение лиц: от тепла к реализму с помощью ИИ
2026-02-13 11:44