Автор: Денис Аветисян
Новая реализация метода iCIPT2 демонстрирует высокую масштабируемость и производительность при расчетах сложных молекулярных систем благодаря оптимизированному MPI-параллелизму.
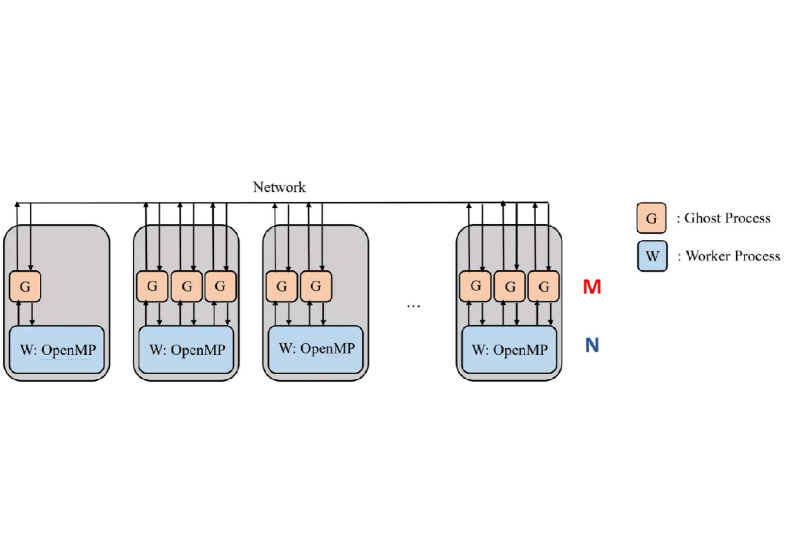
Представлена высокооптимизированная и масштабируемая MPI-реализация метода iCIPT2 для расчетов коррелированных электронных систем.
Вычислительные методы квантовой химии сталкиваются с ограничениями при моделировании сложных молекулярных систем, требующих высокой точности. В статье ‘Unified MPI Parallelization of Wave Function Methods: iCIPT2 as a Showcase’ представлена унифицированная реализация параллелизации на основе MPI для волновых функций, на примере метода iCIPT2, позволяющая абстрагировать каждый вычислительный шаг как динамически планируемый цикл с использованием «призрачных» процессов и последующим глобальным сведением локальных результатов. Достигнутая эффективность параллелизации, достигающая 94% и 89% на 16 узлах (1024 ядрах) для возмущений и полных расчетов соответственно, открывает возможности для исследований больших активных пространств и получения эталонных данных для молекул, таких как циклобутадиен и озон. Не приведет ли дальнейшая оптимизация алгоритмов и расширение возможностей параллелизации к принципиально новым достижениям в моделировании сложных химических процессов?
Основы Точного Молекулярного Моделирования
Точное вычисление электронной структуры является краеугольным камнем для понимания химических явлений, однако представляет собой сложную вычислительную задачу. Моделирование поведения электронов в молекулах, определяющее химические свойства и реакции, требует решения Schrödinger уравнения, сложность которого экспоненциально возрастает с числом электронов. Вследствие этого, даже для относительно небольших молекул, точное решение становится практически невозможным на современных компьютерах. Ученые разрабатывают различные приближенные методы и алгоритмы, направленные на снижение вычислительных затрат, сохраняя при этом приемлемый уровень точности. Эти методы, такие как методы теории функционала плотности или кластерные методы, позволяют исследовать химические процессы, которые иначе оставались бы недоступными для теоретического анализа, однако всегда требуют компромисса между точностью и вычислительной эффективностью.
Традиционные методы, такие как метод Хартри-Фока, несмотря на свою историческую значимость в квантовой химии, часто вынуждены прибегать к приближениям для упрощения сложных расчетов. Эти приближения, направленные на снижение вычислительных затрат, неизбежно влияют на достоверность получаемых результатов. Например, пренебрежение электронной корреляцией, возникающей из-за мгновенного взаимодействия электронов, может приводить к существенным погрешностям в определении энергий и геометрии молекул. Более того, корректное описание динамической корреляции, отражающей мгновенные изменения электронной структуры, представляет особую сложность для однодетерминантных методов, таких как Хартри-Фока. В результате, хотя метод Хартри-Фока и является отправной точкой для многих расчетов, для получения высокоточных результатов часто требуется использование более сложных методов, учитывающих электронную корреляцию, например, метод конфигурационных взаимодействий или теория возмущений.
В расчетах электронной структуры молекул, особенно при рассмотрении тяжелых элементов, необходимо учитывать релятивистские эффекты. Эти эффекты, обусловленные приближением скорости электронов к скорости света, становятся значимыми из-за увеличения массы электронов и, как следствие, уменьшения их длины волны де Бройля. Игнорирование релятивистских поправок приводит к существенным ошибкам в предсказании свойств, таких как энергия ионизации, сродство к электронам и геометрия молекулы. В частности, для элементов, начиная с селена и далее по периодической таблице, релятивистские эффекты приводят к сжатию s— и p-орбиталей, экранированию внешних электронов и, как следствие, изменению химической реакционной способности. Точное моделирование этих эффектов требует использования релятивистских уравнений, таких как уравнение Дирака, что существенно усложняет вычислительные задачи, но необходимо для получения достоверных результатов в химии тяжелых элементов и материаловедении.
iCIPT2: Современный Подход к Теории Волн
Метод iCIPT2 представляет собой итеративный подход к вычислению волновых функций, сочетающий в себе конфигурационную диаграмму взаимодействий (CI) и теорию возмущений. В отличие от чистого CI, который может быть вычислительно дорогим для больших систем, iCIPT2 использует возмущения второго порядка для эффективного учета корреляционных эффектов. Итеративный процесс CI обеспечивает постепенное улучшение приближения к точной волновой функции, а включение теории возмущений стабилизирует вычисления и позволяет достичь высокой точности при умеренных вычислительных затратах. Данный подход обеспечивает сбалансированное сочетание точности и эффективности, что делает его привлекательным для моделирования сложных химических систем.
Метод iCIPT2 использует теорию возмущений второго порядка (second-order perturbation theory) для повышения точности и сходимости расчетов, а также для уменьшения вычислительных затрат. В частности, применение теории возмущений позволяет эффективно учитывать эффекты электронной корреляции, которые не учитываются в методах Хартри-Фока. Для дальнейшей оптимизации вычислений в iCIPT2 используются естественные орбитали (natural orbitals). Естественные орбитали представляют собой линейную комбинацию базисных функций, которые наиболее эффективно описывают электронную плотность, что приводит к более быстрой сходимости и снижению вычислительной сложности за счет исключения наименее важных орбиталей из расчетов.
Приближение замороженного ядра (Frozen-Core Approximation) значительно повышает эффективность расчетов в методе iCIPT2, особенно для систем, где преобладают валентные электроны. Данный подход заключается в исключении основных электронов из корреляционных расчетов, рассматривая их как невозмущаемые. Это существенно снижает размер базисного множества и, следовательно, вычислительные затраты, поскольку корреляционные эффекты, вносимые основными электронами, обычно незначительны для многих химических систем. Практически, это означает, что вычисления проводятся только с участием валентных электронов, что позволяет существенно сократить время расчета и требуемые ресурсы, сохраняя при этом достаточную точность для большинства приложений.
Стратегии Параллелизации для Повышения Производительности
Для эффективного использования современных вычислительных архитектур, программный комплекс iCIPT2 использует параллелизацию на основе технологий OpenMP и MPI. OpenMP применяется для распараллеливания вычислений внутри отдельного узла, позволяя использовать многоядерные процессоры. MPI, в свою очередь, обеспечивает параллельную работу на нескольких узлах кластера, распределяя вычислительную нагрузку между ними. Комбинирование этих двух подходов позволяет iCIPT2 масштабироваться как на локальных многопроцессорных системах, так и на крупных вычислительных кластерах, что критически важно для моделирования сложных молекулярных систем.
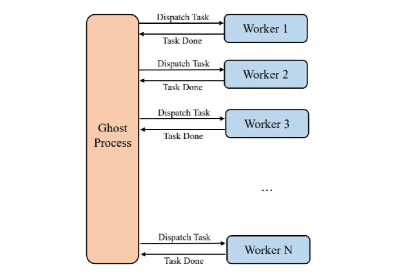
Динамическое распределение задач в iCIPT2 осуществляется посредством специализированного “процесса-призрака”, который контролирует балансировку нагрузки между ядрами процессора и вычислительными узлами. Этот процесс непрерывно отслеживает доступность ресурсов и перераспределяет вычислительные задачи, избегая простоя отдельных ядер или узлов. В отличие от статического распределения, динамический подход позволяет эффективно использовать все доступные ресурсы, особенно при работе с неоднородными вычислительными системами или при возникновении неравномерной нагрузки на различные части расчетной области. Алгоритм динамического планирования адаптируется к изменяющимся условиям, обеспечивая оптимальную производительность даже при колебаниях нагрузки.
Параллельная схема, использующая OpenMP и MPI, демонстрирует эффективность, превышающую 89% при использовании 16 вычислительных узлов. Данный показатель эффективности подтверждается значительным сокращением времени вычислений для сложных молекулярных систем, что позволяет проводить моделирование более масштабных и детализированных структур. Эффективность рассчитывается как отношение времени выполнения последовательной версии кода к времени выполнения параллельной версии, умноженное на количество используемых узлов. Результаты показывают, что увеличение числа узлов до 16 обеспечивает существенное ускорение вычислений при минимальных потерях эффективности, что делает iCIPT2 эффективным инструментом для вычислительной химии и материаловедения.
Области Применения и Значение для Химического Понимания
Методология iCIPT2 продемонстрировала свою эффективность при исследовании процесса автомеризации циклобутадиена — фундаментальной реакции в органической химии. Применение данного подхода позволило детально изучить потенциальную энергию поверхности реакции, выявив ключевые стадии и переходные состояния. Успешное моделирование этого сложного процесса подтверждает применимость iCIPT2 к изучению других реакций, характеризующихся значительными электронными корреляционными эффектами, и открывает перспективы для более точного предсказания и понимания химических превращений.
Полученная поверхность потенциальной энергии позволила детально изучить механизм реакции и характер переходных состояний при автомеризации циклобутадиена. Анализ этой поверхности выявил ключевые этапы перестройки, включая геометрию и энергию переходных состояний, что позволило установить последовательность событий, приводящих к образованию бензола. Детальное представление о форме поверхности потенциальной энергии позволило не только визуализировать реакционный путь, но и количественно оценить энергетические барьеры, определяющие скорость и селективность процесса автомеризации. Это углубленное понимание реакционного механизма стало возможным благодаря точному моделированию электронных взаимодействий и использованию передовых вычислительных методов.
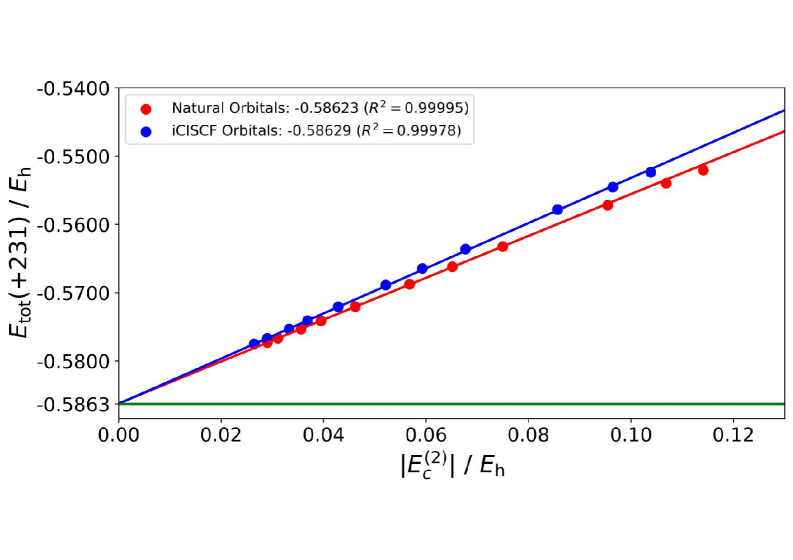
Результаты вычислительных исследований демонстрируют, что барьер автомерного превращения циклобутадиена составляет 8.91 ккал/моль, что находится в превосходном согласии с наиболее точной теоретической оценкой в 8.93 ккал/моль. Наряду с этим, были рассчитаны энергии корреляции для бензола, составившие -864.41 мЭг (cc-pVDZ) и -1034.0 мЭг (cc-pVTZ). Такое соответствие между полученными данными и существующими теоретическими предсказаниями подтверждает надежность и точность использованного методологического подхода, а также позволяет с уверенностью использовать его для дальнейшего изучения сложных химических процессов и уточнения понимания фундаментальных свойств молекул.
![Расчеты для циклобутадиеновой аутомеризации с использованием базисного набора aug-cc-pVTZ показывают, что энергия ошибки <span class="katex-eq" data-katex-display="false">\Delta E = E_{\mathrm{tot}}[C_{\mathrm{min}}] - E_{\mathrm{tot}}[0]</span> линейно зависит от количества CSF (коэффициент детерминации R² = 0.9994, 0.9983, 0.9992, 0.9986) и демонстрирует высокую точность iCIPT2.](https://arxiv.org/html/2602.04470v1/x8.png)
Представленная работа демонстрирует стремление к упрощению сложных вычислений в рамках метода iCIPT2. Авторы эффективно использовали возможности параллелизации MPI, добиваясь высокой производительности при моделировании коррелированных электронных систем. Этот подход к оптимизации, направленный на устранение избыточности, перекликается с принципом, сформулированным Галилео Галилеем: «Природа не заботится о том, чтобы объяснять свои законы». Как и в науке, где необходимо отбросить лишнее, чтобы увидеть суть, так и в данной работе, удаление ненужных сложностей позволяет достичь большей ясности и эффективности в вычислениях. Успешная реализация динамического распределения задач подчеркивает, что истинное понимание достигается не добавлением деталей, а их умелым отбрасыванием.
Что дальше?
Представленная работа, хотя и демонстрирует значительный прогресс в параллелизации метода iCIPT2, не решает фундаментальную проблему: стоимость точных расчетов коррелированных электронных систем. Ускорение вычислений — лишь временное облегчение, подобно уменьшению шума, а не устранению его источника. Поиск алгоритмов, масштабирующихся лучше, чем полиномиально, остается задачей, требующей не столько вычислительной мощи, сколько принципиально нового подхода.
Ограничения текущей реализации, связанные с динамическим распределением задач, предполагают необходимость дальнейшей оптимизации коммуникационных затрат. Однако, истинное упрощение, возможно, лежит не в улучшении существующих методов, а в разработке приближений, позволяющих жертвовать малой частью точности ради существенного снижения вычислительной нагрузки. Погоня за абсолютной точностью, порой, оказывается бесплодной.
Будущие исследования должны сосредоточиться на интеграции представленного подхода с другими методами квантовой химии, а также на разработке адаптивных стратегий, автоматически выбирающих наиболее эффективный метод в зависимости от характеристик исследуемой системы. Иначе, сложность лишь множится, а ясность остается недостижимой.
Оригинал статьи: https://arxiv.org/pdf/2602.04470.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Отражения культуры: Как языковые модели рассказывают истории
- Укрощение Бесконечности: Алгебраические Инструменты для Кватернионов и За их Пределами
- Самообучающиеся агенты: новый подход к автономным системам
- Графы и действия: новый подход к планированию для роботов
- Квантовые маршруты и гравитационные сенсоры: немного иронии от физика
- Квантовые состояния под давлением: сжатие данных для новых алгоритмов
- Визуальное мышление машин: проверка на прочность
- Искусственный разум: Нет доказательств самосознания в современных языковых моделях
- Третья Разновидность ИИ: Как модели, думающие «про себя», оставят позади GPT и CoT
- Квантовые амбиции: Иран вступает в гонку
2026-02-05 11:40