Автор: Денис Аветисян
Новая модель позволяет предсказывать активность генов по изображениям тканей, открывая возможности для более глубокого понимания онкологических процессов.
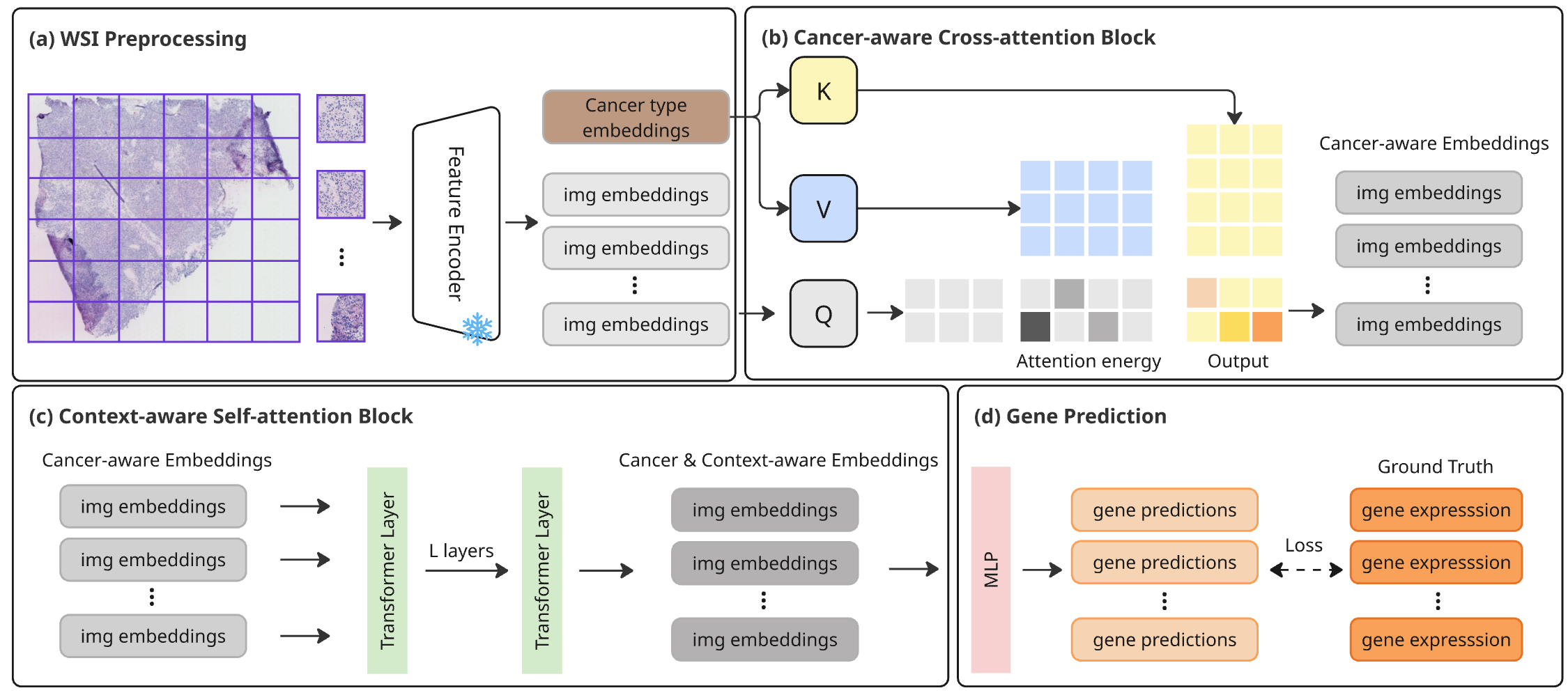
HistoPrism — это трансформерная модель, точно предсказывающая экспрессию генов из гистологических изображений при панканцерном анализе и обеспечивающая биологически релевантную оценку на основе когерентности путей.
Несмотря на растущий интерес к прогнозированию экспрессии генов по данным гистологии, сохраняется проблема обобщения моделей на различные типы рака и оценки биологической значимости полученных результатов. В данной работе, представленной статьей ‘HistoPrism: Unlocking Functional Pathway Analysis from Pan-Cancer Histology via Gene Expression Prediction’, предлагается HistoPrism — эффективная архитектура на основе трансформеров для пан-ракового предсказания экспрессии генов по изображениям гистологии. Модель демонстрирует превосходство над существующими подходами не только в предсказании экспрессии отдельных генов, но, что особенно важно, и в восстановлении когерентных транскриптомных паттернов на уровне функциональных сигнальных путей. Открывает ли это новые перспективы для клинически релевантного моделирования транскриптома на основе рутинно получаемых гистологических препаратов?
Морфология и Геномика: Преодоление Разрыва в Понимании
Традиционный геномный анализ зачастую проводится без учета морфологического контекста, что приводит к упущению важных сведений о механизмах развития заболеваний. Исследования, сосредоточенные исключительно на генетических мутациях, могут не выявить ключевые изменения в структуре тканей и клеточном строении, которые являются прямыми проявлениями болезни. Такое разделение подходов ограничивает понимание сложных взаимодействий между генотипом и фенотипом, особенно в гетерогенных заболеваниях, таких как рак. Недооценка морфологических признаков может привести к неточным диагнозам, неэффективному лечению и затрудняет разработку персонализированных терапевтических стратегий, учитывающих индивидуальные особенности патологического процесса.
Анализ гистопатологических изображений, полученных при гематоксилин-эозиновом окрашивании (H&E), представляет собой значительную проблему из-за их внутренней сложности и высокой размерности. Каждое изображение содержит огромное количество пикселей, каждый из которых несет информацию о цвете и интенсивности, что создает многомерное пространство данных. Выделение биологически значимых сигналов из этого пространства требует сложных алгоритмов, способных отфильтровать шум и идентифицировать паттерны, связанные с конкретными заболеваниями, такими как рак. Проблема усугубляется тем, что эти паттерны часто тонки и могут различаться между разными пациентами и типами опухолей, что требует разработки адаптивных методов анализа изображений, способных учитывать эту гетерогенность.
В онкологических исследованиях остро ощущается потребность в методах, объединяющих визуальную информацию, полученную при анализе гистологических препаратов, и геномные данные. Традиционно эти два типа информации рассматриваются изолированно, что ограничивает понимание сложных механизмов развития рака. Интеграция морфологических признаков, таких как форма и структура опухолевых клеток, с геномными данными, включая мутации и экспрессию генов, позволит создать более полную и точную картину заболевания. Такой подход обещает не только улучшить диагностику и прогнозирование, но и выявить новые терапевтические мишени, адаптированные к индивидуальным характеристикам опухоли. Разработка подобных методов представляет собой важный шаг к персонализированной онкологии, где лечение подбирается с учетом всех аспектов заболевания.
Фундаментальные Модели для Патологии: Новый Подход к Анализу
Патолого-анатомические фундаментальные модели (PFM), такие как UNI PFM и Gigapath PFM, используют масштабные наборы данных для обучения устойчивым визуальным представлениям на основе гистопатологических изображений. Этот подход позволяет моделям выявлять и кодировать важные визуальные характеристики тканей, присутствующие в изображениях, что существенно улучшает их способность к обобщению и адаптации к различным задачам анализа. Объем используемых данных играет критическую роль: чем больше изображений включено в обучающую выборку, тем более надежными и точными становятся полученные визуальные представления, поскольку модель получает возможность изучить широкий спектр морфологических признаков и вариаций.
Предварительное обучение моделей на больших объемах данных изображений гистопатологии позволяет им извлекать релевантные признаки, необходимые для решения различных задач, таких как предсказание экспрессии генов. Этот процесс включает в себя анализ миллионов изображений тканей, что позволяет моделям научиться идентифицировать паттерны и структуры, связанные с различными биологическими процессами. Извлеченные признаки представляют собой векторные представления изображений, которые могут быть использованы в качестве входных данных для алгоритмов машинного обучения, направленных на прогнозирование молекулярных характеристик опухолей, таких как уровень экспрессии определенных генов, что потенциально может улучшить диагностику и выбор терапии.
STPath представляет собой значительный прогресс в области разработки фундаментальных моделей для патологии, являясь пан-раковой моделью, обученной на масштабных данных гистопатологических изображений. Ключевой особенностью STPath является использование подхода маскированного моделирования генов (masked gene modeling), позволяющего модели предсказывать экспрессию генов непосредственно из визуальной информации, полученной с гистологических срезов. Это позволяет связать визуальные характеристики ткани с молекулярными процессами, открывая возможности для более точной диагностики и прогнозирования течения заболеваний.
Оценка Биологической Достоверности: Когерентность Путей Генов
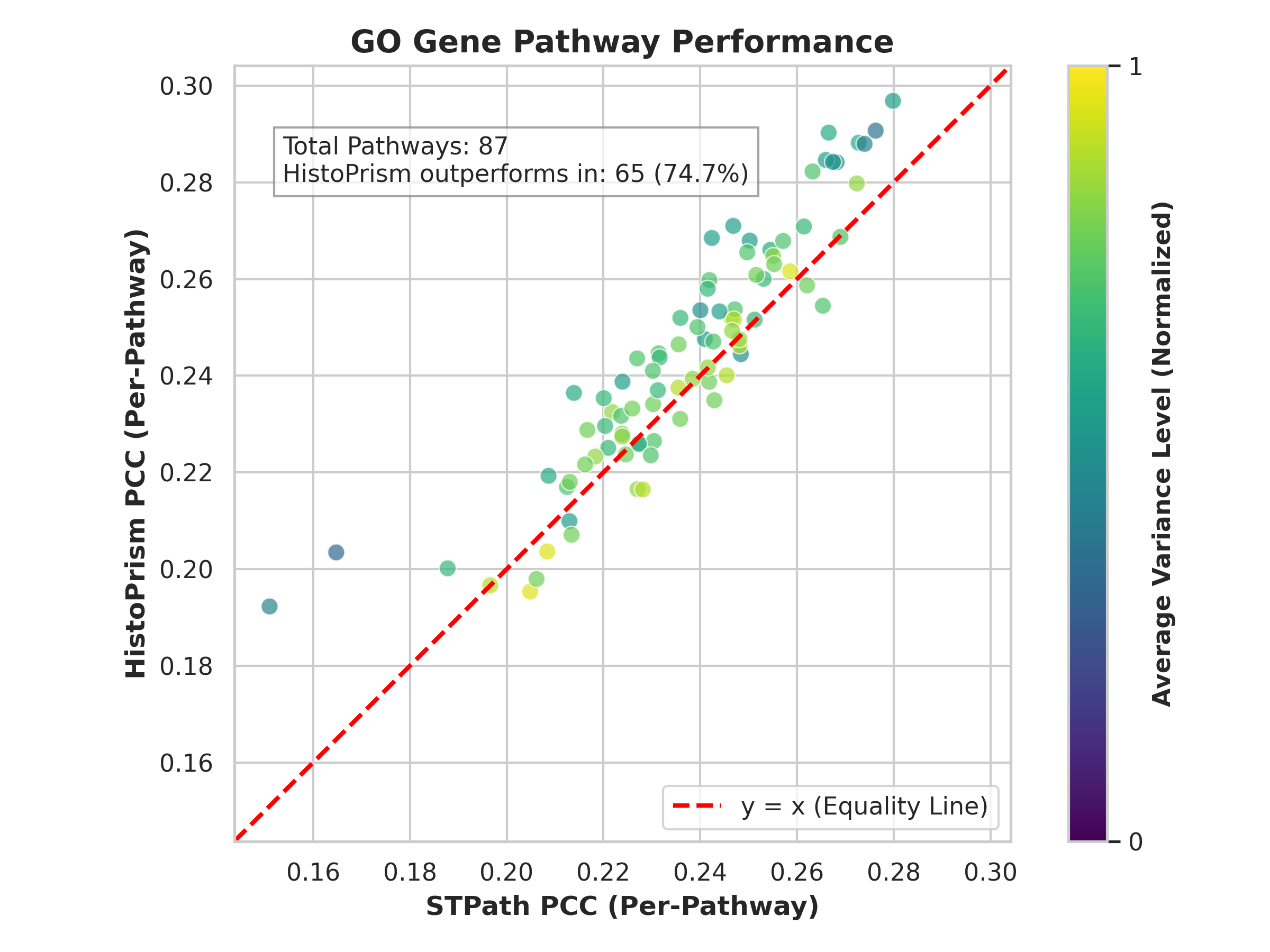
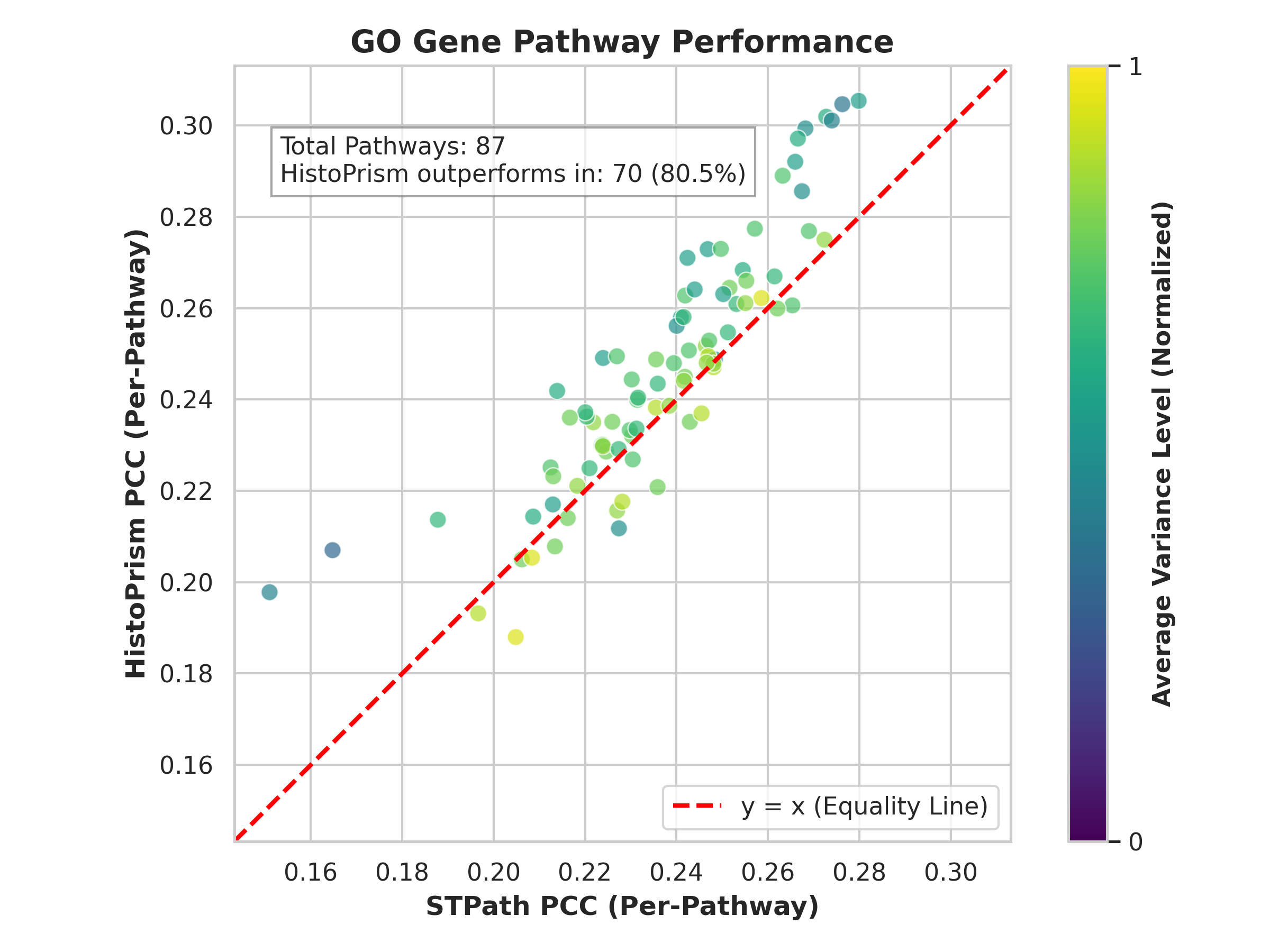
Метод когерентности путей генов (Gene Pathway Coherence, GPC) представляет собой новый подход к оценке биологической значимости предсказанных изменений экспрессии генов. В отличие от традиционных методов, основанных на простой корреляции с эталонными данными, GPC оценивает, насколько предсказанные изменения экспрессии согласуются с известными биологическими процессами, представленными в базах данных Gene Ontology и Hallmark Gene Sets. Это позволяет выявить, соответствуют ли предсказания ожидаемым изменениям в функциональных единицах клетки, а не просто отражают статистическую схожесть с экспериментальными данными. GPC позволяет более точно оценить биологическую релевантность предсказаний, особенно в случаях, когда корреляция с эталонными данными может быть недостаточной для подтверждения значимости наблюдаемых изменений.
Метод когерентности путей генов (Gene Pathway Coherence, GPC) оценивает соответствие предсказанных изменений экспрессии генов с установленными биологическими процессами, представленными в базах данных Gene Ontology Pathways и Hallmark Gene Sets. В рамках GPC, предсказанные изменения экспрессии анализируются на предмет их согласованности с ожидаемыми изменениями в генах, входящих в конкретные биологические пути. Высокая степень когерентности указывает на то, что предсказания отражают биологически значимые изменения, а не случайные корреляции. Анализ проводится на уровне путей, что позволяет оценить, насколько полно и точно предсказания отражают изменения в функционировании биологических систем.
Для количественной оценки когерентности предсказанной экспрессии генов на уровне путей, в рамках Gene Pathway Coherence (GPC), используется корреляция Пирсона с высоковариабельными генами. Данный подход позволяет определить степень соответствия предсказаний известным биологическим процессам, поскольку высоковариабельные гены часто являются ключевыми регуляторами этих процессов. Вычисление корреляции Пирсона для каждого гена в заданном пути и последующее усреднение результатов по всем генам пути обеспечивает надежную метрику, отражающую согласованность предсказаний с ожидаемыми изменениями экспрессии в рамках данного биологического пути. Использование корреляции Пирсона в GPC повышает устойчивость оценки качества предсказаний, минимизируя влияние случайных отклонений и обеспечивая более точную оценку биологической релевантности.
В процессе валидации предсказаний, платформа HistoPrism использует фреймворк Gene Pathway Coherence (GPC), демонстрируя показатель в 86.0% по Hallmark путям и 74.7% по путям Gene Ontology. Данные результаты свидетельствуют о высокой степени соответствия предсказанных изменений экспрессии генов установленным биологическим процессам. При сравнении с платформой STPath, HistoPrism демонстрирует более высокие показатели GPC, что указывает на улучшенную способность платформы к точному моделированию биологических процессов на уровне путей.
Анализ данных показывает, что алгоритм HistoPrism демонстрирует значительно более высокие значения Adjusted Mutual Information (AMI) при глобальной кластеризации по сравнению с алгоритмом STPath. AMI является метрикой, оценивающей сходство между кластеризацией, выполненной алгоритмом, и истинными метками классов, если таковые известны. Более высокое значение AMI указывает на более точную и надежную кластеризацию, подтверждающую, что предсказания HistoPrism лучше соответствуют ожидаемым биологическим структурам и закономерностям, чем предсказания STPath. Данный результат служит дополнительным подтверждением повышенной прогностической силы алгоритма HistoPrism.
Пан-Раковый Анализ и Перспективы Развития
Система HistoPrism, объединенная с методом GPC (Graph-based Path Clustering), предоставляет мощный инструмент для всестороннего пан-ракового анализа. Исследование демонстрирует способность платформы выявлять как общие, так и специфические геномные и транскриптомные характеристики, присущие различным типам рака. Такой подход позволяет не только глубже понять фундаментальные механизмы развития онкологических заболеваний, но и способствует обнаружению потенциальных биомаркеров, общих для нескольких видов рака. Выявление общих закономерностей в геномном профиле различных онкологий открывает перспективы для разработки универсальных терапевтических стратегий, а идентификация уникальных особенностей каждого типа рака позволяет адаптировать лечение, повышая его эффективность и снижая побочные эффекты. Анализ, проведенный с помощью HistoPrism и GPC, предоставляет бесценную информацию для дальнейших исследований в области онкологии и разработки персонализированной медицины.
В архитектуре HistoPrism ключевую роль играет механизм кросс-внимания, позволяющий эффективно объединять визуальные данные, полученные при анализе гистологических изображений, с геномной информацией. Этот подход позволяет модели сопоставлять морфологические характеристики опухоли с её генетическим профилем, выявляя взаимосвязи, которые ранее оставались незамеченными. В результате, точность прогнозирования различных параметров, таких как ответ на терапию или вероятность рецидива, значительно повышается. Кросс-внимание позволяет HistoPrism фокусироваться на наиболее релевантных участках изображения и генома, игнорируя шум и нерелевантную информацию, что делает анализ более надежным и эффективным.
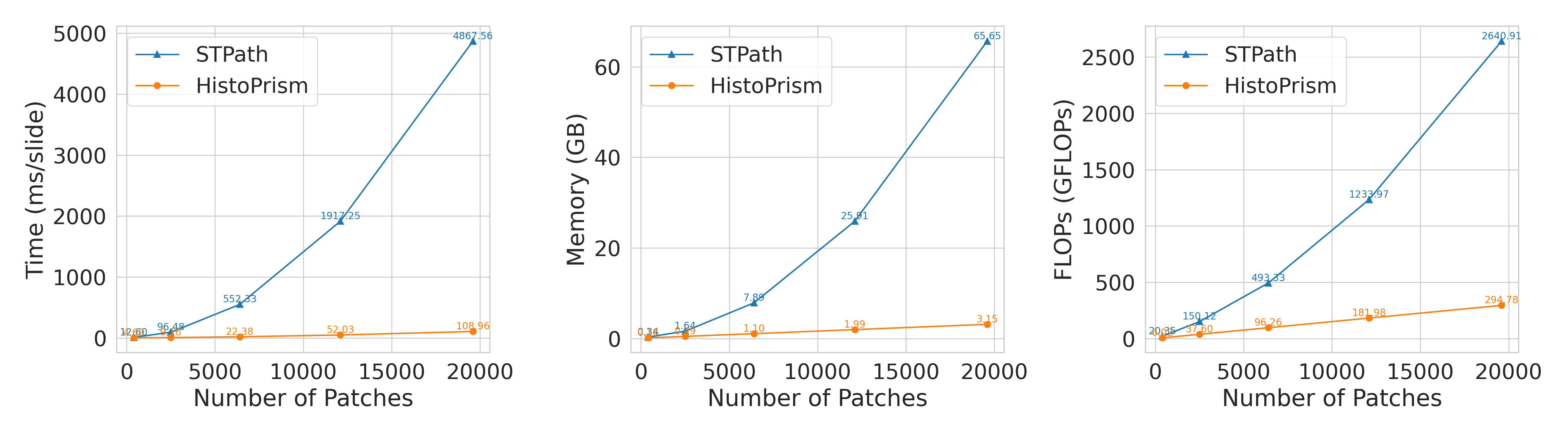
Разработанная платформа HistoPrism демонстрирует значительное повышение вычислительной эффективности по сравнению с существующей системой STPath. Исследования показали, что HistoPrism требует существенно меньше пиковой памяти GPU и значительно снижает количество операций с плавающей запятой (FLOPs) при выполнении аналогичных задач. Данное улучшение позволяет анализировать более крупные и сложные наборы данных, используя менее мощное оборудование, а также сокращает время, необходимое для получения результатов. В отличие от STPath, демонстрирующего экспоненциальный рост вычислительных затрат с увеличением объема данных, HistoPrism масштабируется линейно, что делает её особенно перспективной для применения в масштабных пан-раковых исследованиях и клинической практике.
Исследования показали, что HistoPrism демонстрирует принципиально иную масштабируемость по сравнению с существующими методами, такими как STPath. В то время как вычислительные затраты STPath растут экспоненциально с увеличением объема анализируемых данных, HistoPrism сохраняет линейную зависимость. Это означает, что даже при значительном увеличении размера датасета, требуемые вычислительные ресурсы для HistoPrism растут предсказуемо и умеренно. Такая эффективность позволяет анализировать гораздо большие объемы данных, открывая возможности для всестороннего изучения раковых заболеваний и повышения точности прогнозов, что особенно важно для разработки персонализированных стратегий лечения.
Достижения в области анализа данных, такие как разработанная платформа HistoPrism, открывают новую эру в персонализированной медицине. Теперь врачи получают возможность разрабатывать индивидуальные стратегии лечения, основываясь на уникальном генетическом профиле и морфологических особенностях каждого пациента. Это означает, что выбор терапии будет учитывать не только тип рака, но и конкретные мутации, экспрессию генов и визуальные характеристики опухоли, что значительно повышает вероятность успешного лечения и снижает риск побочных эффектов. Подобный подход позволяет отойти от универсальных схем лечения и перейти к терапии, максимально адаптированной к индивидуальным потребностям организма, что является ключевым шагом к повышению эффективности борьбы с онкологическими заболеваниями.
Дальнейшие исследования направлены на интеграцию пространственной транскриптомики, что позволит существенно повысить точность прогнозов и углубить понимание микроокружения опухоли. Использование данных о пространственном распределении экспрессии генов предоставит возможность более детально изучить взаимодействие между опухолевыми клетками и окружающими тканями, выявить ключевые факторы, влияющие на прогрессирование заболевания, и определить специфические мишени для терапии. Ожидается, что объединение морфологических данных, полученных с помощью HistoPrism, с информацией о пространственной экспрессии генов позволит создать комплексные модели, отражающие гетерогенность опухоли и предсказывающие ее реакцию на различные виды лечения, открывая новые перспективы для персонализированной онкологической помощи.
Представленная работа демонстрирует стремление к математической чистоте в анализе данных, что находит отражение в подходе к предсказанию экспрессии генов по изображениям гистологии. Модель HistoPrism, основанная на архитектуре Transformer, стремится не просто к высокой точности предсказаний, но и к биологической релевантности, оцениваемой через когерентность путей. Как однажды заметил Давид Гильберт: «В математике нет спектра. Есть только математика.» Эта фраза подчеркивает, что истинная ценность заключается не в приближенных решениях, а в строгой доказательности и универсальности подхода. Подобно этому, HistoPrism стремится к созданию фундаментальной модели, способной обобщать знания между различными типами рака, что является проявлением стремления к математической элегантности и универсальности в биоинформатике.
Куда двигаться дальше?
Представленная работа, несомненно, демонстрирует впечатляющую способность модели HistoPrism к экстраполяции информации о генной экспрессии из гистологических изображений. Однако, необходимо признать, что корреляция — не причина. Модель успешно предсказывает, но не объясняет лежащие в основе биологические механизмы, связывающие морфологию с генной активностью. Использование когерентности путей — шаг в правильном направлении, но остается лишь прокси-метрикой биологической релевантности. Истинное понимание потребует интеграции с другими модальностями данных и разработки методов, способных выявлять причинно-следственные связи.
Очевидным ограничением является зависимость от существующих данных о генной экспрессии для обучения. Модель, по сути, воспроизводит известные закономерности, а не открывает принципиально новые. Будущие исследования должны быть направлены на разработку методов обучения без учителя или с частичным учителем, способных извлекать знания непосредственно из гистологических изображений, минуя необходимость в «золотом стандарте» данных о генной экспрессии. Иначе это лишь элегантная интерполяция, а не настоящая научная революция.
Наконец, необходимо критически оценить устойчивость модели к артефактам, возникающим при подготовке образцов и окрашивании. Любая кажущаяся биологическая значимость, основанная на таких артефактах, — это иллюзия, а не открытие. Строгий контроль качества и разработка методов, устойчивых к таким искажениям, — задача, требующая пристального внимания. Иначе, элегантный алгоритм станет лишь инструментом для увековечивания ошибок.
Оригинал статьи: https://arxiv.org/pdf/2601.21560.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Отражения культуры: Как языковые модели рассказывают истории
- Квантовые Заметки: Прогресс и Парадоксы
- Звуковая фабрика: искусственный интеллект, создающий музыку и речь
- Квантовый оптимизатор: Новый подход к сложным задачам
- Кванты в Финансах: Не Шутка!
- Гармония в коде: Распознавание аккордов с помощью глубокого обучения
- Взлом языковых моделей: эволюция атак, а не подсказок
- Квантовый скачок из Андхра-Прадеш: что это значит?
- Квантовые симуляторы: точное вычисление энергии основного состояния
- Искусственный интеллект на допросе: как объяснить решения в цифровой криминалистике?
2026-02-01 06:49