Автор: Денис Аветисян
Новое исследование раскрывает внутренние механизмы ESMFold, объясняя, как эта модель искусственного интеллекта моделирует сложную структуру белков.
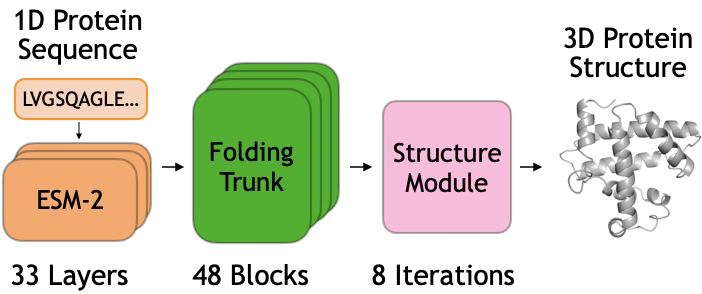
Исследователи выявили двухэтапный процесс в ESMFold: ранние блоки обрабатывают биохимическую информацию, а поздние — уточняют геометрические связи, что позволяет лучше понять принципы предсказания структуры белков.
Несмотря на впечатляющие успехи в предсказании структуры белков, механизмы, лежащие в основе работы моделей искусственного интеллекта, остаются малоизученными. В работе ‘Mechanisms of AI Protein Folding in ESMFold’ исследуется, как модель ESMFold «сворачивает» белки, выявляя двухэтапный процесс: на ранних этапах происходит инициализация парных биохимических сигналов, а на поздних — уточнение пространственных взаимосвязей. Полученные результаты позволяют локализовать и отследить механизмы принятия решений моделью, манипулируя ими с сильным причинно-следственным эффектом. Сможем ли мы, понимая эти внутренние процессы, создать еще более эффективные и интерпретируемые модели предсказания структуры белков?
Раскрытие Кода Сворачивания: От Последовательности к Структуре
Предсказание трехмерной структуры белка на основе его аминокислотной последовательности остаётся одной из ключевых задач вычислительной биологии. Сложность заключается в огромном количестве возможных конформаций, которые белок может принимать, и необходимости точного определения наиболее стабильной из них. Несмотря на десятилетия исследований и разработок, точное предсказание структуры, необходимое для понимания функции белка и разработки новых лекарственных препаратов, до сих пор представляет собой значительную проблему. Успешное решение этой задачи позволит не только расширить знания о фундаментальных процессах жизни, но и откроет новые горизонты в области биотехнологий и медицины, значительно ускорив разработку новых методов лечения различных заболеваний.
Традиционные методы предсказания структуры белков сталкиваются с колоссальными трудностями, обусловленными сложностью энергетического ландшафта и огромным разнообразием возможных конформаций. Каждая молекула белка обладает бесчисленным множеством способов «сворачивания», и поиск наиболее стабильной структуры требует учета множества слабых взаимодействий. Проблема усугубляется тем, что энергетические различия между этими конформациями часто крайне малы, что делает поиск оптимальной структуры сравнимым с поиском иголки в стоге сена. Вычислительные ресурсы, необходимые для полного исследования всего конформационного пространства, быстро становятся непосильными, даже для самых мощных суперкомпьютеров, что подталкивает исследователей к разработке инновационных подходов, способных эффективно преодолеть эти ограничения.
Новый подход, представленный моделью ESMFold, существенно отличается от традиционных методов предсказания структуры белка. Вместо поиска минимальной энергии в огромном пространстве конформаций, ESMFold использует глубокое обучение для непосредственного предсказания трехмерных координат атомов. Этот метод, анализируя парные характеристики аминокислотных остатков, достигает точности до 70% в определении их идентичности, что открывает новые возможности для понимания связи между последовательностью аминокислот и пространственной структурой белка. Такая высокая точность позволяет значительно ускорить процесс структурного моделирования и предсказания функций белков, представляя собой прорыв в области вычислительной биологии.
Складывающийся Ствол: Уточнение Взаимосвязей
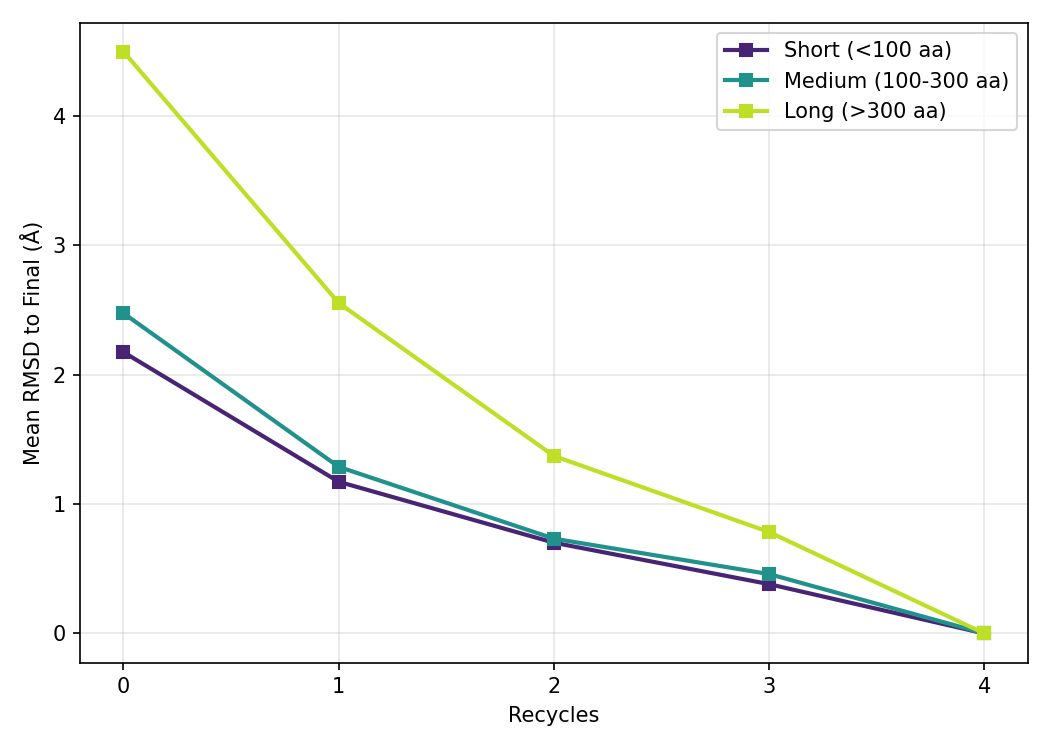
“Складывающийся ствол” (Folding Trunk) представляет собой итеративный процесс, в ходе которого одновременно уточняются как последовательное (sequence) представление белка, так и парное (pairwise) представление, отражающее пространственные взаимосвязи между аминокислотными остатками. Каждая итерация включает в себя взаимное улучшение этих двух представлений, что позволяет модели ESMFold последовательно приближаться к наиболее вероятной трехмерной структуре. Итерации продолжаются до достижения сходимости, обеспечивая высокую точность предсказания структуры белка за счет постепенного уточнения как информации о последовательности, так и информации о пространственном расположении остатков.
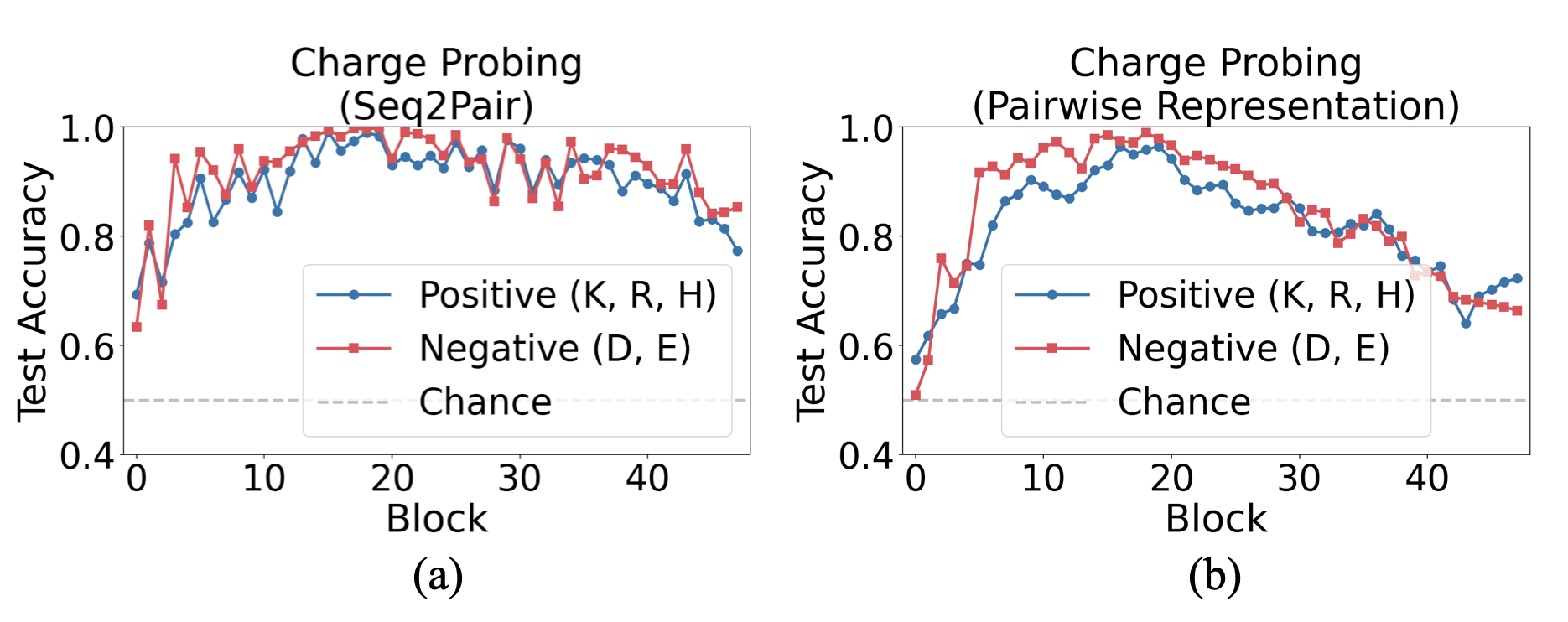
В архитектуре ESMFold обмен информацией между последовательным и парным представлениями белка осуществляется посредством двух основных путей: ‘Sequence-to-Pair’ (Seq2Pair) и ‘Pairwise-to-Sequence’ (Pair2Seq). Seq2Pair преобразует информацию о последовательности аминокислот в парное представление, кодирующее расстояния между остатками. В свою очередь, Pair2Seq выполняет обратное преобразование, возвращая информацию из парного представления в последовательное. Эти итеративные циклы обмена данными позволяют модели уточнять как последовательное, так и парное представления, улучшая точность предсказания структуры белка. В процессе работы информация последовательно передается между этими представлениями, обеспечивая взаимное обогащение данными и повышение качества предсказания.
Итеративный процесс, используемый в ESMFold, позволяет учитывать дальнодействующие зависимости, критически важные для точного сворачивания белка. Обмен информацией между последовательностью и парными представлениями протеина происходит поэтапно, что способствует улавливанию сложных взаимодействий между удалёнными аминокислотными остатками. Полная передача информации о последовательности достигается к блоку 15, после чего модель способна эффективно прогнозировать трёхмерную структуру, опираясь на учёт всех значимых взаимосвязей в белковой цепи.
“Парное представление” в ESMFold функционирует как “карта расстояний”, кодирующая пространственные взаимосвязи между аминокислотными остатками. Это достигается путем хранения информации о взаимном расстоянии между каждой парой остатков в структуре белка. Каждый элемент в парном представлении соответствует расстоянию между конкретными остатками, обеспечивая основу для моделирования трехмерной структуры белка и выявления важных взаимодействий, определяющих его функциональность. Данное представление позволяет алгоритму учитывать не только локальные, но и дальнодействующие связи между остатками, что критически важно для точного предсказания структуры.
От Представления к Геометрии: Модуль Структуры
Модуль «Структура» выполняет преобразование уточненной последовательности и парных представлений в трехмерные координаты. Этот процесс является ключевым этапом в определении пространственного расположения элементов, кодируемых входными данными. Входные данные, состоящие из последовательности и информации о взаимодействиях между элементами, обрабатываются для вычисления x, y, z координат каждого элемента в трехмерном пространстве. Полученные координаты формируют основу для дальнейшего анализа и визуализации структуры, обеспечивая возможность представления данных в понятном и интерпретируемом формате.
Модуль ‘Структура’ использует механизм ‘Инвариантного Точечного Внимания’ (Invariant Point Attention) для определения ключевых пространственных взаимосвязей при предсказании 3D-координат. Этот механизм позволяет модели фокусироваться на наиболее значимых точках и их относительной позиции в пространстве, что критически важно для построения корректной трехмерной структуры. В процессе предсказания координат, ‘Инвариантное Точечное Внимание’ анализирует взаимосвязи между точками, игнорируя инвариантные преобразования, такие как вращения и трансляции, что обеспечивает стабильность и точность предсказываемой структуры. Этот подход позволяет более эффективно использовать информацию, закодированную в последовательности и парных представлениях, для формирования правдоподобной трехмерной конфигурации.
Для оценки качества предсказанных структур используется метрика — радиус гирации R_g. Данный параметр характеризует компактность молекулярной структуры и рассчитывается как среднеквадратичный радиус атомов относительно центра масс. Для исключения неспецифической, чрезмерной компактизации, устанавливается пороговое значение в 0.7. Структуры, демонстрирующие радиус гирации ниже этого порога, отбрасываются, поскольку свидетельствуют о нереалистичной или артефактной компактности, не соответствующей физическим свойствам исследуемых молекул.
Для оценки доступности информации, закодированной в представлениях, используются методы, такие как ‘Linear Probe’. Данный подход позволяет проверить, насколько эффективно представления отражают линейные расстояния между атомами в молекуле. Экспериментальные результаты показали, что коэффициент детерминации R^2 для линейных distance probes достигает значения 0.9, что свидетельствует о высокой степени соответствия между предсказанными представлениями и фактическими геометрическими данными. Это указывает на то, что модуль эффективно кодирует информацию о пространственном расположении атомов, что необходимо для последующего построения трехмерной структуры.
Раскладывая Механизм: Активационный Патчинг и Бета-Петли
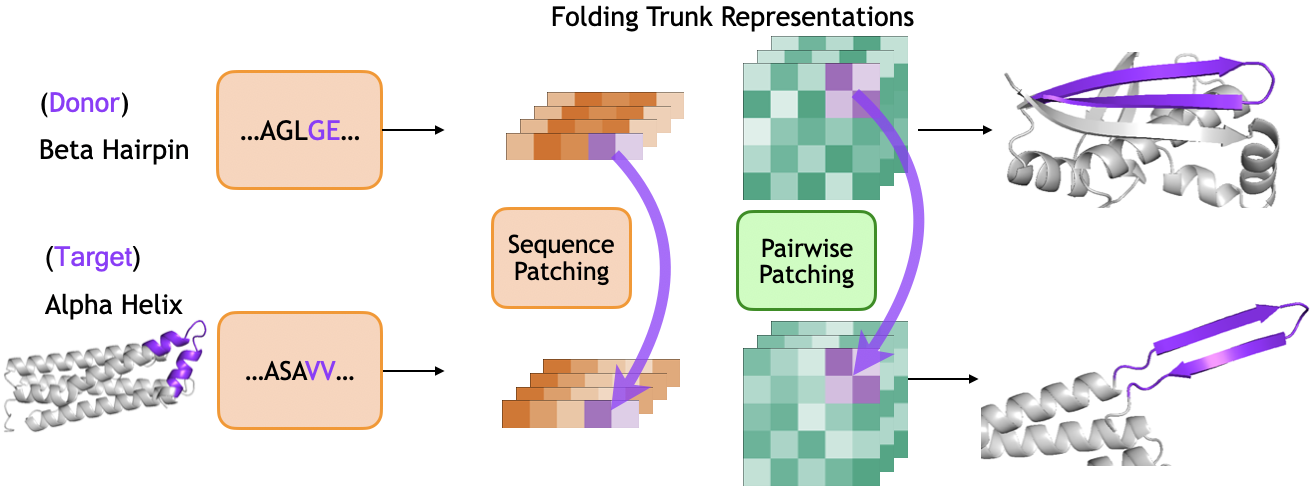
Метод “активационного патчинга” открывает новые возможности для изучения механизмов сворачивания белков, позволяя исследователям переносить структурные представления между различными белками. Этот подход не ограничивается простым наблюдением за процессом, а дает возможность установить причинно-следственные связи между отдельными элементами структуры и конечным результатом — правильно сложенной молекулой. Фактически, ученые могут искусственно “внедрить” определенные характеристики одного белка в другой, и оценить, как это повлияет на его способность к сворачиванию. Успешное применение патчинга демонстрирует, что модели машинного обучения, такие как ESMFold, способны не просто запоминать известные структуры, но и действительно понимать принципы, определяющие процесс фолдинга, открывая путь к разработке белков с заданными свойствами.
Исследование укладки простых мотивов, таких как бета-петли, позволяет ученым проверить, насколько глубоко модель понимает фундаментальные принципы формирования структуры белков. Изучение этих относительно простых структур служит своеобразным «лакмусовой бумажкой», демонстрируя, способна ли модель предсказывать укладку белков, основываясь на физических принципах, а не просто запоминая существующие структуры. Успешное моделирование укладки бета-петель, с предсказуемыми взаимодействиями между аминокислотами, подтверждает, что модель действительно выучила важные закономерности, определяющие стабильность и формирование белковых структур. Этот подход позволяет отделить истинное понимание принципов укладки от простого запоминания данных, что является ключевым шагом в разработке более надежных и точных методов предсказания структуры белков.
Исследования бета-петлей продемонстрировали, что их стабильность напрямую определяется электростатической комплементарностью между аминокислотными остатками. Модель ESMFold, анализируя взаимодействие зарядов и распределение потенциалов на поверхности белка, способна точно предсказывать и воспроизводить эти ключевые взаимодействия, обеспечивая устойчивость структуры. Данный результат подчеркивает, что ESMFold не просто запоминает известные структуры, но и активно усваивает фундаментальные принципы, управляющие формированием белковых комплексов, что подтверждается высокой точностью моделирования даже для простых, но важных мотивов, таких как бета-петли.
Анализ причинно-следственных связей показал, что ESMFold не просто запоминает структуры белков, а активно изучает принципы их формирования. Успешное применение метода «активационного патчинга» — перенос представлений между белками для анализа механизмов сворачивания — привело к формированию бета-петли в 40% случаев. Этот результат свидетельствует о способности модели к генерации новых структур на основе усвоенных принципов, а не просто к воспроизведению ранее увиденных, и подтверждает, что ESMFold действительно понимает фундаментальные закономерности, определяющие процесс сворачивания белков.
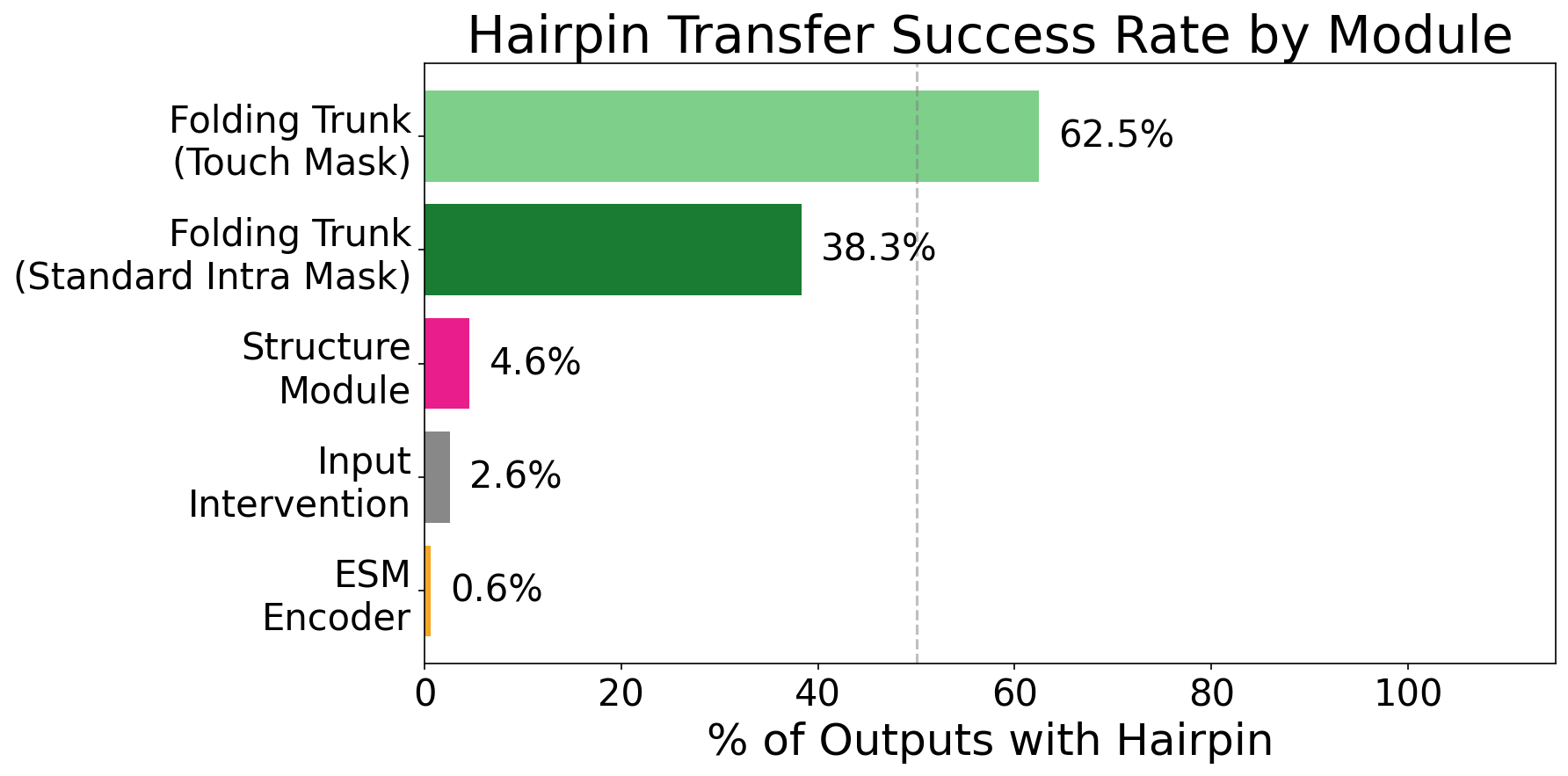
Исследование механизмов работы ESMFold демонстрирует, что предсказание структуры белка — это не просто статистическая задача, а последовательный процесс извлечения и уточнения информации. Ранние блоки нейронной сети, как показывает анализ, служат для передачи базовых биохимических свойств, в то время как поздние блоки отвечают за геометрическую точность. Этот двухступенчатый подход подтверждает, что даже в сложных нейронных сетях можно выделить логичные этапы обработки данных. Как заметил Кен Томпсон: «В конечном счете, все сводится к простоте, и если вы не можете объяснить что-то просто, значит, вы сами этого не понимаете». Эта фраза отражает суть данной работы: стремление к пониманию внутренних механизмов ESMFold, чтобы не просто получить результат, а объяснить, как он достигается, и выявить математическую чистоту алгоритма.
Что Дальше?
Представленное исследование, проливая свет на внутреннюю работу ESMFold, лишь подчеркивает фундаментальную проблему: понимание как нейронная сеть достигает результата не равнозначно пониманию самого процесса сворачивания белка. Наблюдаемое разделение на ранние и поздние блоки — это, безусловно, полезное эмпирическое открытие, однако оно требует строгой математической формализации. Необходимо доказать, что именно эти блоки ответственны за передачу биохимической информации и уточнение геометрических связей, а не просто констатировать этот факт на основе наблюдений. Оптимизация без анализа — это самообман и ловушка для неосторожного разработчика.
Будущие исследования должны сосредоточиться на разработке методов, позволяющих не просто интерпретировать активации сети, но и предсказывать их поведение на основе аминокислотной последовательности. Использование методов активационного патчинга — перспективный шаг, но он требует расширения и формализации. Ключевым вопросом остается, возможно ли создание модели, которая не просто предсказывает структуру, но и объясняет, почему белок сворачивается именно таким образом. До тех пор, пока мы не сможем вывести законы, управляющие этим процессом, любые улучшения в точности предсказания останутся лишь техническими усовершенствованиями, лишенными глубокого научного смысла.
В конечном итоге, истинная элегантность в этой области проявится не в создании все более сложных нейронных сетей, а в разработке простых, понятных и доказуемых алгоритмов, способных воспроизвести фундаментальные принципы сворачивания белков. Именно к этому и следует стремиться, избегая соблазна бесконечной оптимизации «черных ящиков».
Оригинал статьи: https://arxiv.org/pdf/2602.06020.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Укрощение Бесконечности: Алгебраические Инструменты для Кватернионов и За их Пределами
- Самообучающиеся агенты: новый подход к автономным системам
- Визуальный след: Сжатие рассуждений для мощных языковых моделей
- Bibby AI: Новый помощник для исследователей в LaTeX
- Графы и действия: новый подход к планированию для роботов
- Квантовые хроники: Последние новости в области квантовых исследований и разработки.
- Стиль сквозь века: математика искусства
- Самообучающийся Автопилот: Новый Подход к Безопасности и Адаптации
- Управление Формой: Новый Метод Контроля 3D-Генерации
- Гармония в коде: Распознавание аккордов с помощью глубокого обучения
2026-02-07 02:31