Автор: Денис Аветисян
Новый обзор демонстрирует, как детальное описание химических связей позволяет существенно повысить точность прогнозирования свойств материалов с помощью машинного обучения.
Критический анализ квантово-химических дескрипторов связей для предсказания теплопроводности и других свойств материалов, а также их применение в машинном обучении и символической регрессии.
Несмотря на значительные успехи в машинном обучении для материаловедения, учет химической связи как ключевого фактора, определяющего свойства материалов, остается сложной задачей. В работе, озаглавленной ‘A critical assessment of bonding descriptors for predicting materials properties’, проведена критическая оценка дескрипторов, основанных на характере химической связи, для предсказания свойств материалов. Разработанный на основе квантово-химического анализа набор дескрипторов продемонстрировал улучшение предсказательной силы моделей машинного обучения для упругих, колебательных и термодинамических свойств. Возможно ли создание более интерпретируемых и точных моделей материаловедения за счет систематического включения информации о химической связи и ее влиянии на свойства материалов?
За пределами состава: Необходимость дескрипторов связи
Традиционные дескрипторы материалов, такие как те, что используются в MATMINER, зачастую оказываются недостаточно чувствительными для фиксации тонких различий в химических связях, что критически важно для точного предсказания свойств. Эти дескрипторы, основанные преимущественно на составе материала, не способны адекватно отразить нюансы электронной структуры, определяющие, например, ковалентный, ионный или металлический характер связи. В результате, даже небольшие изменения в типе или силе химической связи могут оставаться незамеченными, приводя к неточностям в прогнозировании ключевых характеристик, таких как теплопроводность или механическая прочность. Неспособность уловить эти тонкости ограничивает возможности моделирования материалов и поиска новых соединений с заданными свойствами, подчеркивая необходимость разработки более совершенных дескрипторов, непосредственно учитывающих природу химической связи.
Прогнозирование свойств материалов напрямую зависит от понимания лежащих в основе химических связей, однако существующие методы зачастую оказываются неспособными адекватно отразить их сложность. Традиционные подходы, фокусирующиеся на элементарном составе, игнорируют тонкие различия в электронном строении, которые определяют, как атомы взаимодействуют друг с другом. Например, два материала с одинаковым составом могут демонстрировать совершенно разные характеристики, если их химические связи организованы по-разному. Это связано с тем, что свойства материалов — будь то электропроводность, теплопроводность или механическая прочность — являются результатом не только того, из чего сделан материал, но и как эти компоненты связаны между собой. Поэтому для точного прогнозирования необходимы более совершенные инструменты, способные учитывать не только состав, но и структуру и характер химических связей.
Для точного описания материалов и прогнозирования их свойств необходимо выйти за рамки простого определения состава. Традиционные дескрипторы, основанные исключительно на элементарном составе, не способны уловить тонкие различия в химической связи, определяющие поведение материала. Вместо этого, требуется разработка дескрипторов, которые напрямую количественно оценивают электронную структуру вещества — распределение электронов и характер связей между атомами. Такой подход позволит учитывать ковалентный, ионный, металлический и другие типы связи, а также степень их локализации или делокализации, что, в свою очередь, существенно повысит точность предсказания свойств, таких как теплопроводность, механическая прочность и электрическая проводимость. Использование дескрипторов, основанных на электронной структуре, открывает путь к созданию более реалистичных моделей и, как следствие, к разработке материалов с заданными характеристиками.
Ограничения существующих методов описания материалов существенно затрудняют точное предсказание важных свойств, таких как теплопроводность и механическая прочность. Несмотря на постоянные усилия по совершенствованию алгоритмов, прирост точности предсказаний остается незначительным, составляя в среднем лишь 2-3%. Это указывает на необходимость принципиально нового подхода к описанию материалов, который учитывал бы не только их состав, но и тонкости химической связи. Существующие дескрипторы, основанные исключительно на элементном составе, недостаточно чувствительны для выявления этих нюансов, что и является причиной низкой эффективности современных прогностических моделей. Повышение точности предсказаний требует разработки дескрипторов, непосредственно отражающих электронную структуру и характер взаимодействия между атомами в материале.
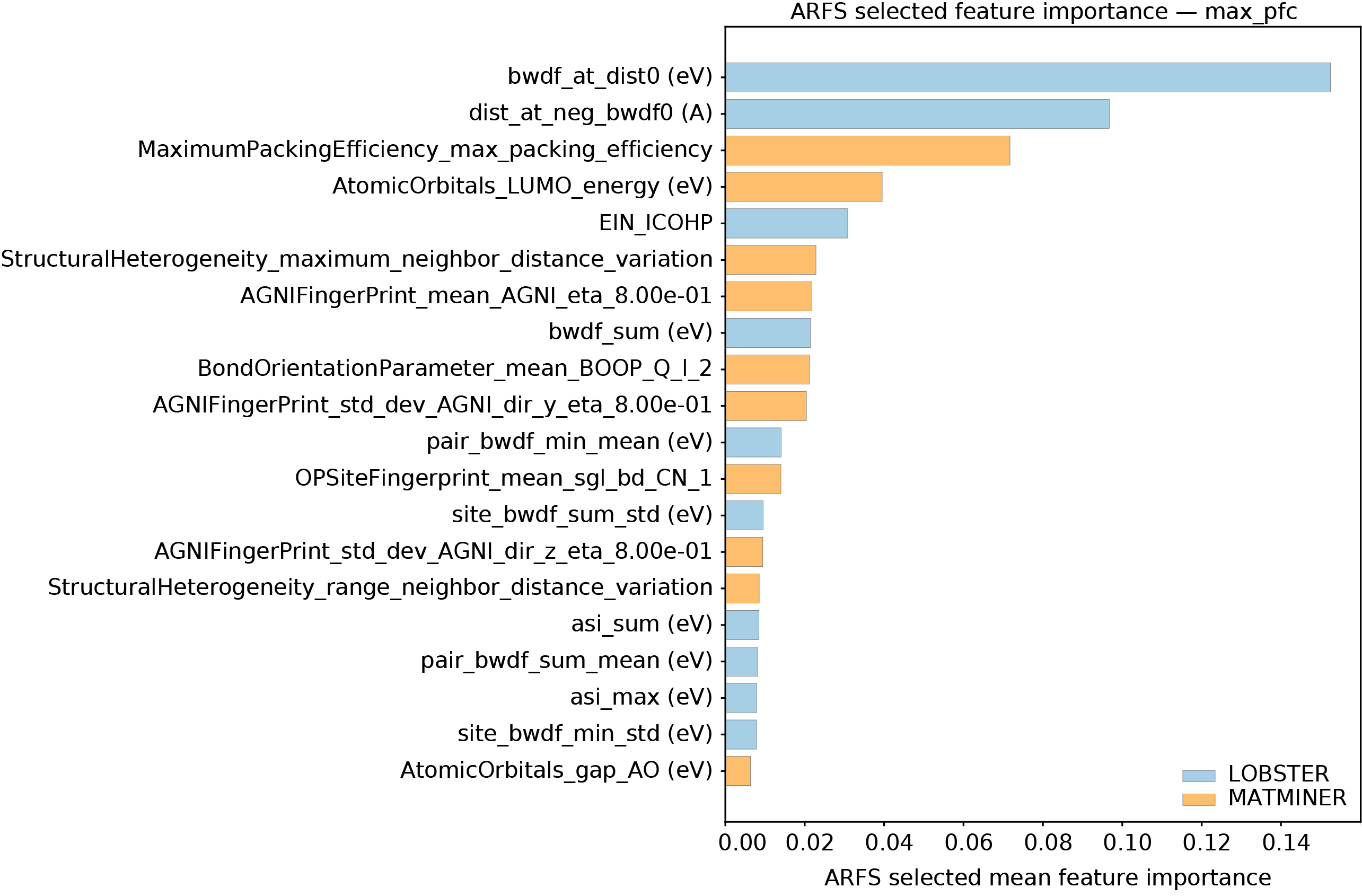
Квантово-химическая основа: Определение дескрипторов связи
Дескрипторы связей, получаемые на основе квантово-химических расчетов, предоставляют эффективный инструмент для характеризации электронной структуры и природы химических связей в материалах. В отличие от традиционных подходов, основанных на анализе валентных связей или электроотрицательности, эти дескрипторы позволяют количественно оценить вклад различных орбиталей в формирование связи, определить ее ионный или ковалентный характер, а также выявить слабые взаимодействия, существенно влияющие на свойства материала. Расчет дескрипторов связей требует решения уравнения Шрёдингера для рассматриваемой системы, что позволяет получить детальное описание распределения электронной плотности и энергии, а также получить информацию о силе и природе связей между атомами. Эти данные необходимы для понимания и прогнозирования физических и химических свойств материалов, включая их стабильность, реакционную способность и оптические характеристики.
Программа LOBSTER является основным вычислительным инструментом для определения квантово-химических дескрипторов связи. Она реализует эффективные алгоритмы для анализа кристаллической орбитальной перекрывки (COOP), плотности состояний связи (COHP) и индекса связи (COBI). LOBSTER позволяет производить расчеты для различных типов химических связей, включая ковалентные, ионные и металлические, а также для систем с комплексной электронной структурой. Программа оптимизирована для работы с большими системами и обеспечивает высокую точность и скорость вычислений, что делает ее незаменимым инструментом для исследований в области материаловедения и химии.
Ключевые дескрипторы, такие как COOP (Crystal Orbital Overlap and Polarization), COHP (Crystal Orbital Hamiltonian Population) и COBI (Crystal Orbital Bond Index), количественно оценивают различные аспекты прочности и характера химической связи. COOP представляет собой меру перекрытия и поляризации кристальных орбиталей, отражая вклад каждой орбитали в общую связь. COHP определяет энергию связи, суммируя вклад орбиталей, связанных с ковалентной связью, и позволяет оценить прочность и характер связи. COBI является интегральным показателем, который измеряет общую степень связи между двумя атомами, учитывая как ковалентный, так и ионный вклад. Совместное использование этих дескрипторов обеспечивает комплексное понимание электронной структуры и характера химических связей в материалах.
Традиционные методы анализа химической связи, такие как рассмотрение типов связей (ковалентная, ионная, металлическая) и суммарного числа валентных электронов, часто оказываются недостаточными для полного описания электронной структуры и влияния её на свойства материалов. В отличие от них, дескрипторы связи, вычисляемые на основе квантово-химических расчетов (COOP, COHP, COBI), позволяют оценить не только наличие связи, но и её силу, характер (например, вклад ковалентной и ионной составляющих) и распределение электронной плотности. Это позволяет учесть тонкие различия в электронной структуре, которые могут существенно влиять на такие свойства материалов, как проводимость, магнитные свойства, оптические характеристики и стабильность.
Машинное обучение для прогнозирования свойств материалов
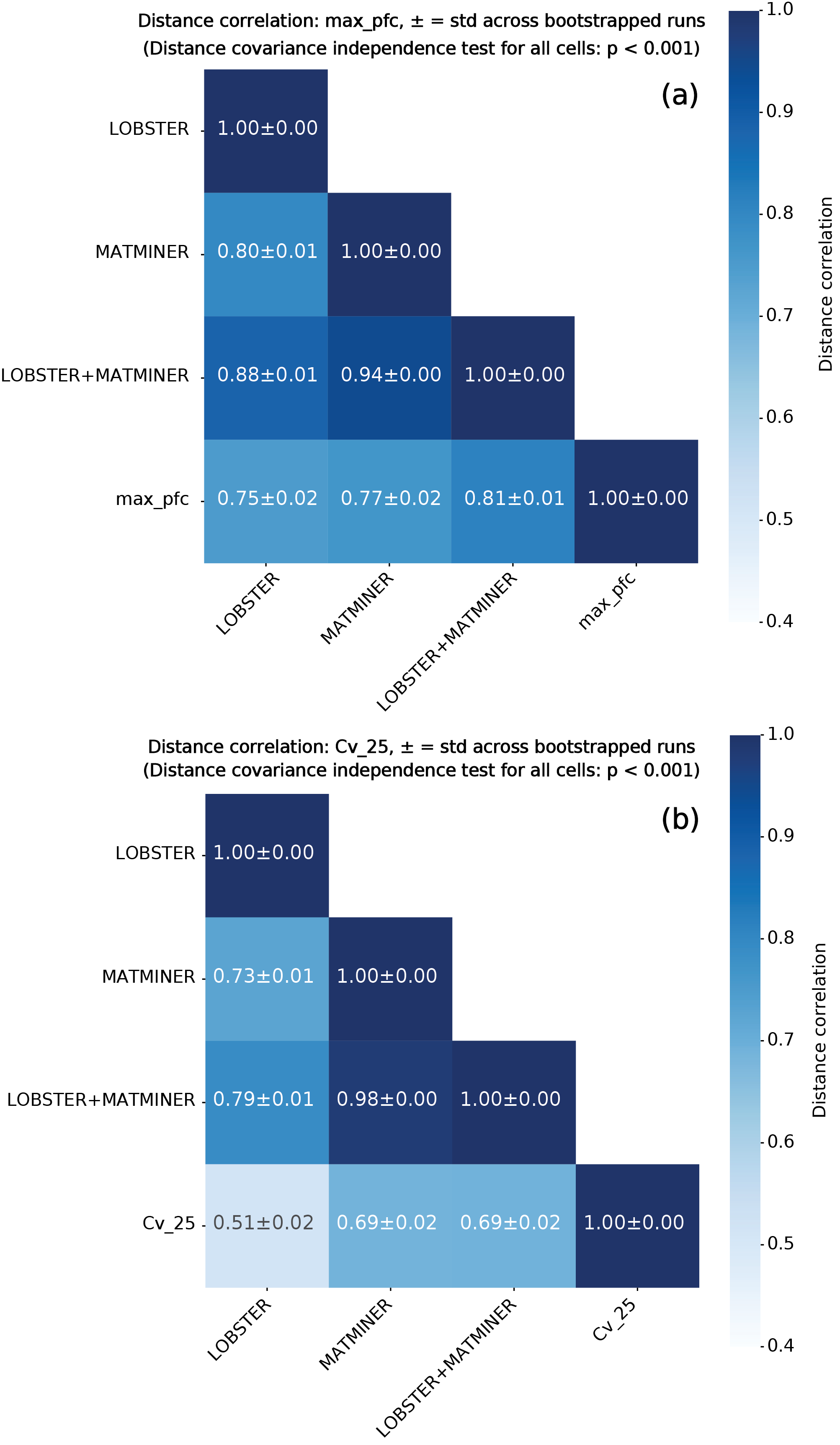
Для предсказания свойств материалов используются модели машинного обучения, в частности, алгоритмы Random Forest и MODNet. Эти модели обучаются на наборе дескрипторов, характеризующих химические связи в материале, и позволяют установить количественную связь между микроскопическими характеристиками связей и макроскопическими свойствами материала. Random Forest, являясь ансамблевым методом, обеспечивает высокую точность и устойчивость предсказаний, а MODNet, использующий нейронные сети, позволяет выявлять более сложные зависимости между дескрипторами и свойствами. Обучение моделей происходит на основе экспериментальных данных о свойствах различных материалов, что позволяет оценивать их прогностическую способность и точность.
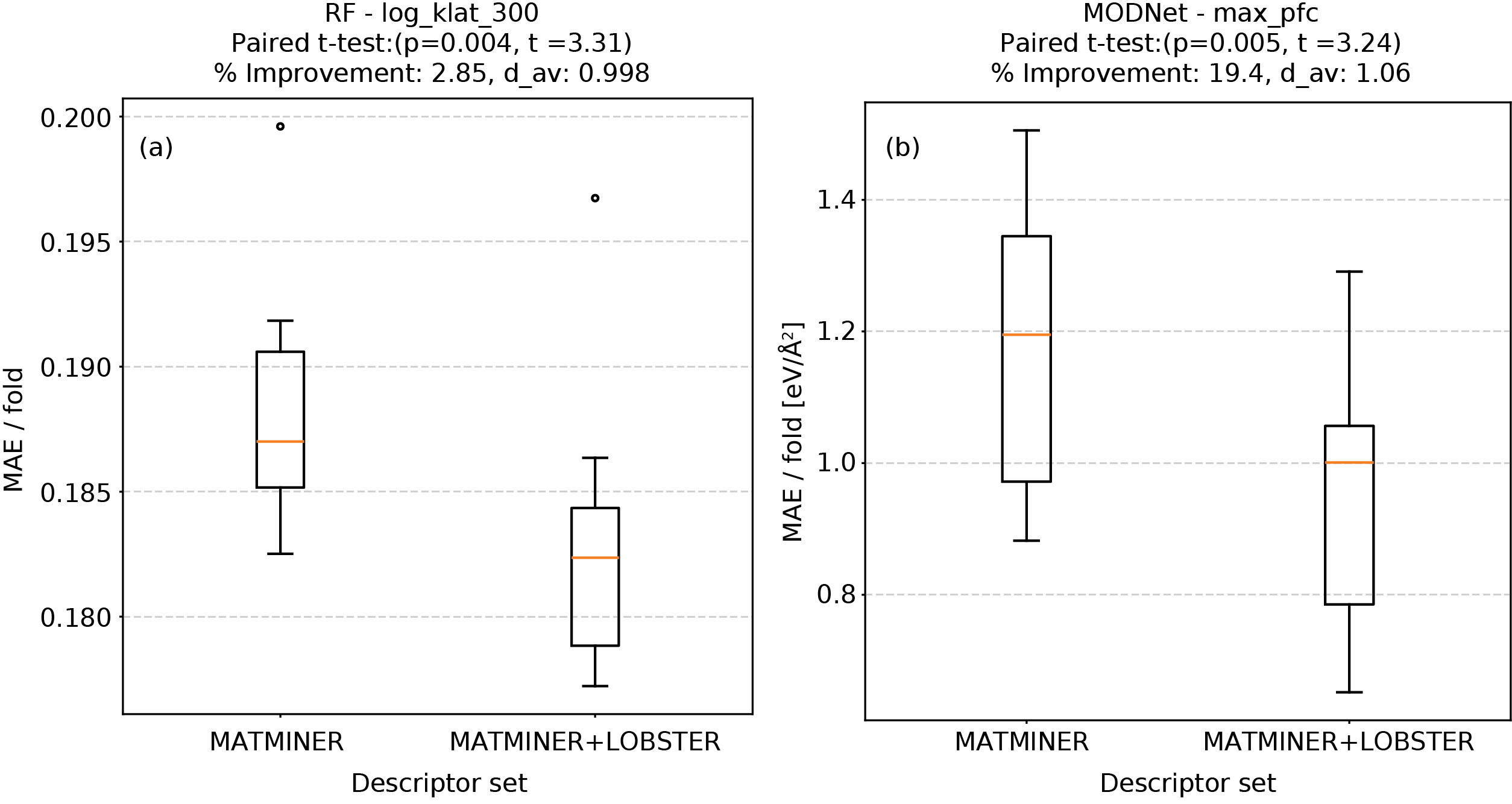
Для повышения эффективности моделей машинного обучения, предсказывающих свойства материалов, применяется метод отбора релевантных признаков (All-Relevant Feature Selection). Данный метод позволяет выделить наиболее информативные подмножества дескрипторов связей, уменьшая размерность входных данных и, как следствие, снижая вычислительную сложность и риск переобучения моделей. Отбор признаков осуществляется на основе оценки их вклада в предсказание целевых свойств материала, что приводит к улучшению обобщающей способности и повышению точности предсказаний. Использование отбора признаков позволяет сконцентрироваться на наиболее значимых характеристиках химической связи, игнорируя избыточную или нерелевантную информацию.
Модели машинного обучения были обучены и протестированы на наборах данных, содержащих информацию о свойствах материалов. Результаты демонстрируют способность моделей точно предсказывать такие параметры, как максимальная постоянная силы, проецируемая на связь K_{max}. В ходе валидации было достигнуто снижение средней абсолютной ошибки (MAE) примерно на 19% по сравнению с существующими методами, что подтверждает эффективность предложенного подхода к прогнозированию свойств материалов на основе характеристик химических связей.
Использование подхода, основанного на анализе данных, позволяет установить устойчивые корреляции между характеристиками химической связи и свойствами материалов. Анализ большого объема данных о свойствах материалов и дескрипторах связей позволяет выявить закономерности, которые не всегда очевидны из традиционных теоретических моделей. Это дает возможность предсказывать свойства материалов, такие как константа максимальной силы, спроецированной на связь K_{max}, на основе их химического состава и структуры. Установленные взаимосвязи позволяют целенаправленно изменять характеристики связей для получения материалов с заданными свойствами, что имеет важное значение для материаловедения и инженерии.
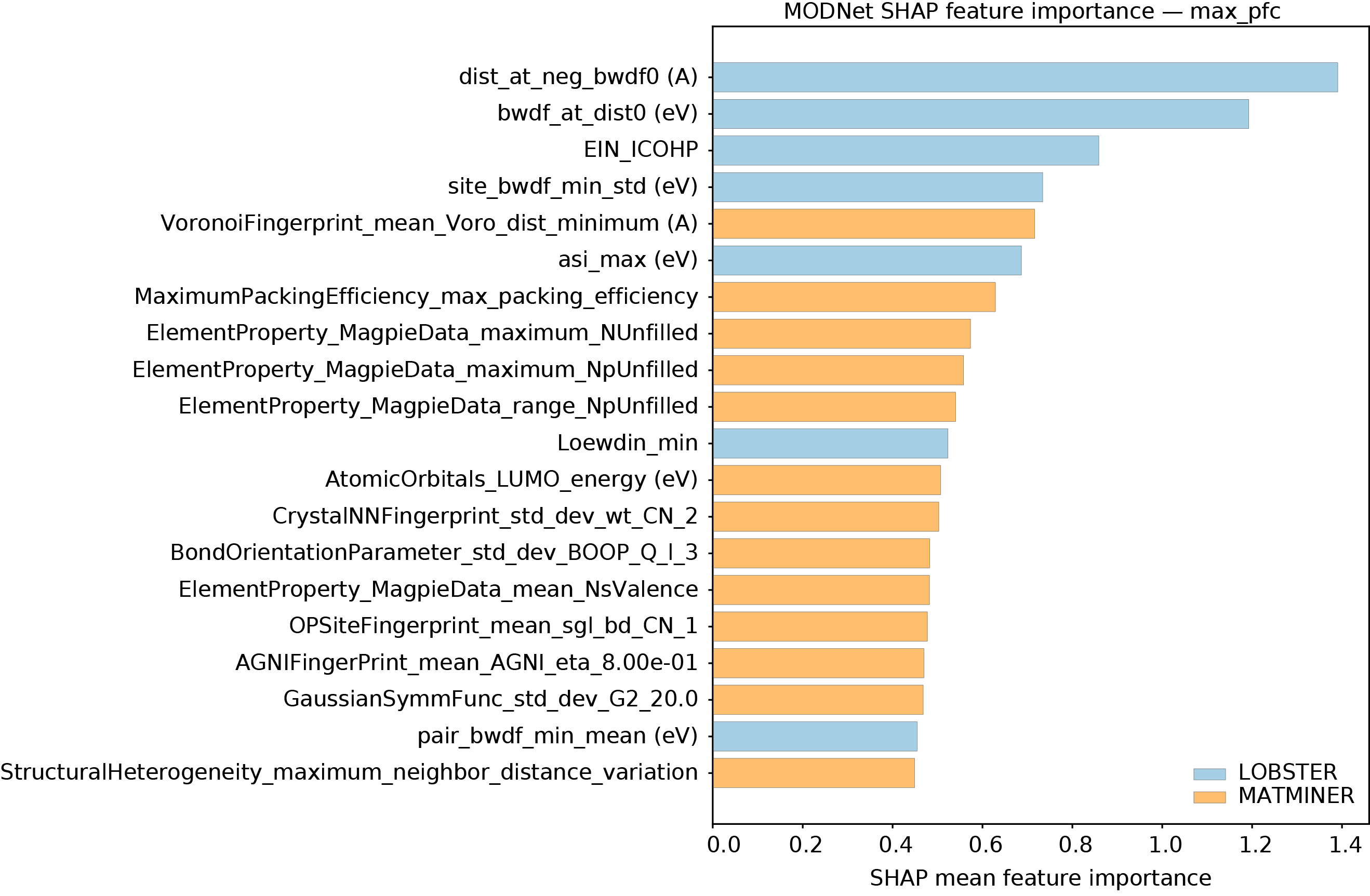
Интерпретируемые выводы: Раскрытие взаимосвязей между связью и свойством
Для выявления взаимосвязей между характеристиками химической связи и свойствами материалов был применен метод символьной регрессии, дополненный методикой SISSO. Этот подход позволил получить аналитические выражения, описывающие зависимость материальных свойств от дескрипторов, характеризующих химическую связь. В отличие от моделей, предсказывающих свойства, данный метод раскрывает фундаментальные механизмы, определяющие поведение материала. Полученные уравнения демонстрируют, какие именно характеристики связи оказывают наибольшее влияние на такие параметры, как модуль сдвига и объемный модуль, предоставляя возможность не просто предсказывать, но и понимать природу этих свойств на основе фундаментальных принципов.
Применение символической регрессии, расширенной методом SISSO, позволяет выйти за рамки простого предсказания свойств материалов и раскрыть лежащие в их основе механизмы. В отличие от моделей, ориентированных исключительно на точность прогноза, данный подход выявляет фундаментальные связи между характеристиками химической связи и наблюдаемыми свойствами. В результате получаются не просто корреляции, а интерпретируемые математические выражения, демонстрирующие, как конкретные аспекты связи — например, log(klat_{300}) с нормализованным ICOHP и log(klat_{300}) с дескриптором, идентифицированным SISSO — влияют на такие параметры, как модуль сдвига и объемный модуль (с коэффициентом корреляции Пирсона r = 0.71 для обоих). Такое понимание принципов, управляющих поведением материала, открывает возможности для целенаправленной разработки новых материалов с заданными характеристиками, опираясь на фундаментальные законы химической связи.
Полученные математические уравнения выявили ключевые характеристики химической связи, оказывающие наибольшее влияние на такие свойства материалов, как модуль сдвига и объемный модуль. Анализ показал высокую корреляцию Пирсона (r = 0.71) между логарифмом klat_{300} и нормализованным ICOHP, а также между klat_{300} и дескриптором, идентифицированным методом SISSO. Это свидетельствует о том, что предложенный подход позволяет не только предсказывать свойства материалов, но и устанавливать прямую связь между особенностями их химической связи и наблюдаемыми характеристиками, что открывает новые возможности для целенаправленного дизайна материалов с заданными свойствами.
Открытие взаимосвязей между характеристиками химической связи и свойствами материалов открывает принципиально новые возможности для целенаправленного дизайна новых соединений. Вместо эмпирического подхода, основанного на переборе различных составов, теперь становится возможным прогнозировать и оптимизировать свойства материалов, исходя из фундаментальных принципов их химической связи. Полученные интерпретируемые модели позволяют выявить, какие конкретно характеристики связи — например, ковалентность, ионность или металлический характер — оказывают наибольшее влияние на такие параметры, как модуль сдвига или объемный модуль. Такой подход не только углубляет понимание природы материаловедения, но и позволяет создавать материалы с заданными свойствами, направленно изменяя их химическую структуру и, следовательно, характеристики связи.
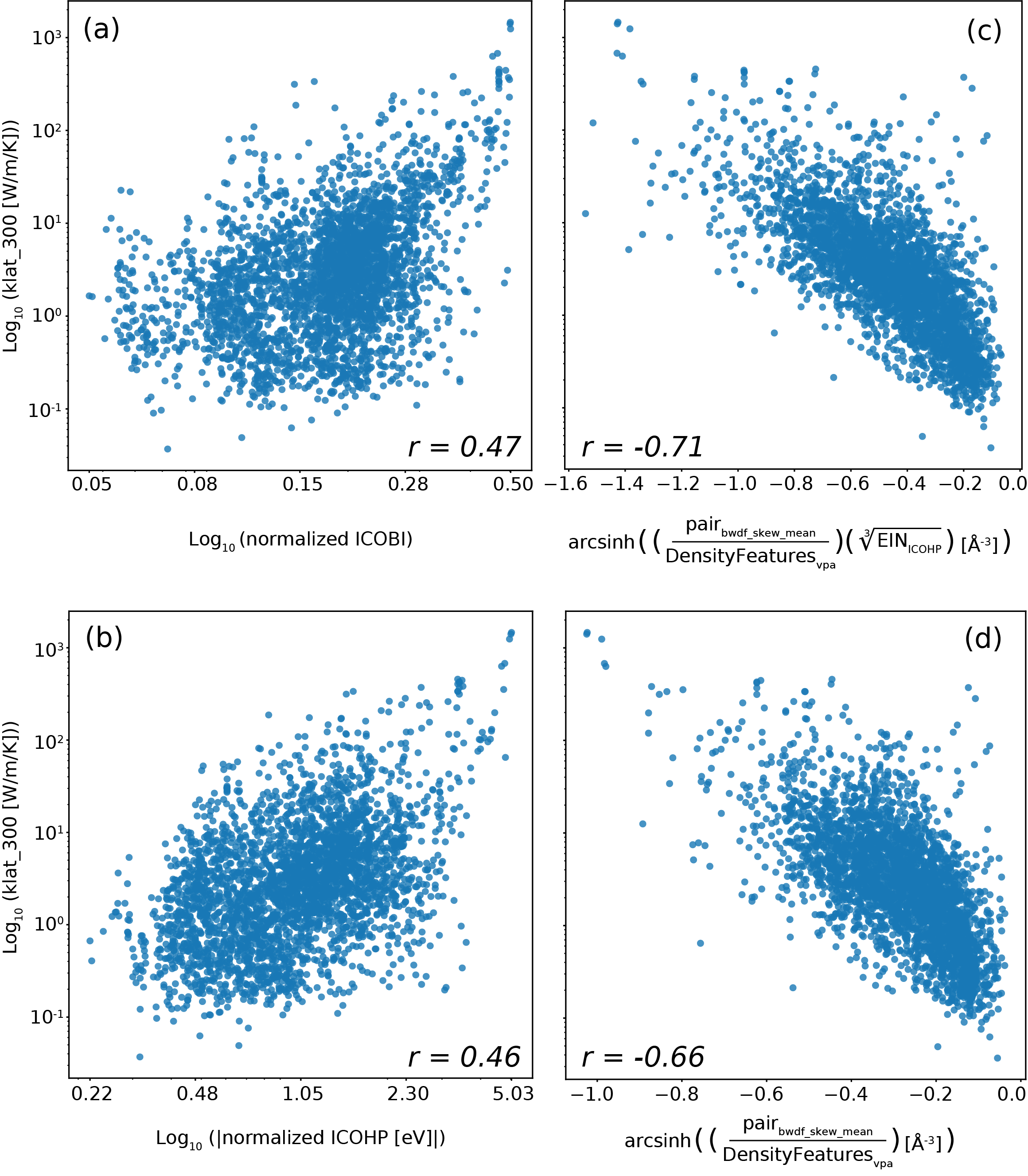
Исследование демонстрирует, что квантово-химические дескрипторы связи значительно повышают предсказательную силу моделей машинного обучения, особенно применительно к свойствам материалов, напрямую зависящим от характера химических связей. Это подтверждает, что любая модель — это моральный акт, поскольку она отражает не только математические зависимости, но и мировоззрение, заложенное в выборе дескрипторов и алгоритмов. Как сказал Людвиг Витгенштейн: «Границы моего языка — границы моего мира». В данном контексте, границы предсказательной способности модели определяются качеством и осмысленностью используемых дескрипторов, отражающих фундаментальные аспекты строения и свойств вещества. Данные выступают зеркалом, а алгоритмы — кистью художника, создающего картину взаимосвязей между структурой и свойствами материалов.
Куда дальше?
Представленная работа демонстрирует, что квантово-химические дескрипторы связей способны повысить прогностическую силу моделей машинного обучения для определённых свойств материалов, особенно тех, что напрямую связаны с характером химических связей. Однако, триумф алгоритмов не должен заслонять собой фундаментальный вопрос: что именно мы автоматизируем? Данные сами по себе нейтральны, но модели отражают предвзятости людей, проектирующих их. Улучшение точности предсказаний без критического осмысления базовых предпосылок — это ускорение без направления.
Очевидной задачей является расширение набора исследуемых свойств и материалов. Но более глубокая проблема заключается в преодолении ограничения, когда дескрипторы становятся лишь «чёрным ящиком», скрывающим физические механизмы. Необходимо разрабатывать инструменты, позволяющие не просто предсказывать свойства, но и понимать, почему материал обладает теми или иными характеристиками. Инструменты без ценностей — это оружие, и ответственность за их применение лежит на тех, кто их создаёт.
В будущем, возможно, следует обратить внимание на разработку дескрипторов, учитывающих не только химические связи, но и более сложные аспекты структуры материала, такие как дефекты и неоднородности. И, что особенно важно, необходимо помнить, что прогресс без этики — это лишь иллюзия движения вперёд.
Оригинал статьи: https://arxiv.org/pdf/2602.12109.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Самообучающиеся агенты: извлечение навыков из открытого кода
- Квантовые Иллюзии и Практические Шаги
- Геном под контролем: Ускорение анализа данных для персонализированной медицины
- Понимание ориентации объектов: новый взгляд на 3D-пространство
- Самообучающиеся автомобили: новый эталон для ИИ-систем
- Масштабная Интерпретация: Новый Взгляд на Надежность Нейросетей
- Искусственный интеллект на службе физики высоких энергий
- Укрощение Бесконечности: Алгебраические Инструменты для Кватернионов и За их Пределами
- Взрыв скорости: Оптимизация внимания для современных GPU
- Геометрические квантовые гейты: на пути к устойчивости
2026-02-13 16:47