Автор: Денис Аветисян
Новый метод вычисления градиентов ядер в рамках ph-AFQMC открывает возможности для высокоточного моделирования молекулярной геометрии и поиска переходных состояний.
Исследование представляет эффективный подход к вычислению ядерных градиентов с использованием автоматического дифференцирования и машинного обучения в контексте phaseless auxiliary-field quantum Monte Carlo.
Вычисление точных ядерных градиентов в рамках методов квантовой Монте-Карло остается сложной задачей, ограничивающей применение этих методов для оптимизации геометрии и поиска переходных состояний. В настоящей работе, посвященной ‘Nuclear gradients from auxiliary-field quantum Monte Carlo and their application in geometry optimization and transition state search’, предложен эффективный подход к вычислению ядерных градиентов в рамках безфазовой вспомогательной квантовой Монте-Карло (ph-AFQMC) с использованием автоматического дифференцирования. Разработанный метод, в сочетании с обучением машинного обучения потенциалов, позволяет успешно оптимизировать геометрию молекул и находить переходные состояния, например, для таутомеризации формимидной кислоты, с результатами, согласующимися с расчетами по методу связанных кластеров. Открывает ли это путь к проведению высокоточных расчетов молекулярной динамики и реакционных путей, недостижимых ранее?
Предел Точности: Вызов Поверхностям Потенциальной Энергии
Точность молекулярного моделирования напрямую зависит от прецизионности потенциальных энергетических поверхностей (ПЭП), однако методы ab initio, обеспечивающие высокую точность, требуют значительных вычислительных ресурсов. Это обстоятельство накладывает существенные ограничения на размер исследуемых систем и временные масштабы моделирования. Несмотря на то, что методы ab initio позволяют рассчитывать энергии взаимодействия атомов с высокой степенью достоверности, их экспоненциальная сложность не позволяет эффективно применять их к системам, состоящим из большого числа атомов или требующим длительного моделирования динамики. Таким образом, поиск компромисса между точностью и вычислительной эффективностью является ключевой задачей в области молекулярного моделирования, что стимулирует разработку новых алгоритмов и приближений для расчета ПЭП.
Традиционные методы, такие как теория функционала плотности (DFT), несмотря на свою вычислительную эффективность, зачастую демонстрируют недостаточную точность при моделировании систем с сильной корреляцией электронов или протекающих химических процессов, требующих детального описания электронного строения. Это связано с тем, что DFT использует приближения для учета многих электронных взаимодействий, что может приводить к существенным погрешностям в расчете энергии и свойств молекул, особенно в случаях, когда электроны сильно взаимодействуют друг с другом или когда необходимо точно описать разрыв химических связей. Например, при изучении переходных металлов, катализаторов или процессов фотосинтеза, где важна корреляция электронов, стандартные функционалы DFT могут давать неверные результаты, что затрудняет предсказание свойств материалов и протекания химических реакций. Для достижения более высокой точности требуется использование более сложных, но и более ресурсоемких методов, учитывающих электронную корреляцию более точно.
Ограничения, связанные с вычислительной сложностью построения точных поверхностей потенциальной энергии, существенно замедляют прогресс в ключевых областях науки и техники. Разработка новых материалов с заданными свойствами, поиск эффективных лекарственных препаратов и детальное изучение химических кинетических процессов требуют моделирования, которое было бы одновременно точным и доступным по вычислительным ресурсам. Затруднения в решении этой задачи препятствуют быстрому скринингу потенциальных кандидатов в материалы, ограничивают возможности предсказания эффективности лекарств и замедляют понимание сложных химических реакций на молекулярном уровне. В связи с этим, активно разрабатываются инновационные подходы, направленные на преодоление этого вычислительного барьера и ускорение научных открытий в указанных областях.
Машинное Обучение Силовых Полей: Путь к Эффективности
Машинное обучение силовых полей (MLFF) представляет собой перспективное решение для снижения вычислительных затрат при моделировании потенциальных энергетических поверхностей (ПЭС). Вместо прямых, ресурсоемких расчетов ab initio, MLFF используют обученные модели для аппроксимации ПЭС. Это достигается путем обучения моделей на данных, полученных из точных, но дорогостоящих расчетов, что позволяет значительно ускорить симуляции молекулярной динамики и другие расчеты, требующие многократного вычисления энергии системы. Эффективность MLFF заключается в возможности прогнозирования энергии системы с приемлемой точностью, используя значительно меньше вычислительных ресурсов, чем традиционные методы.
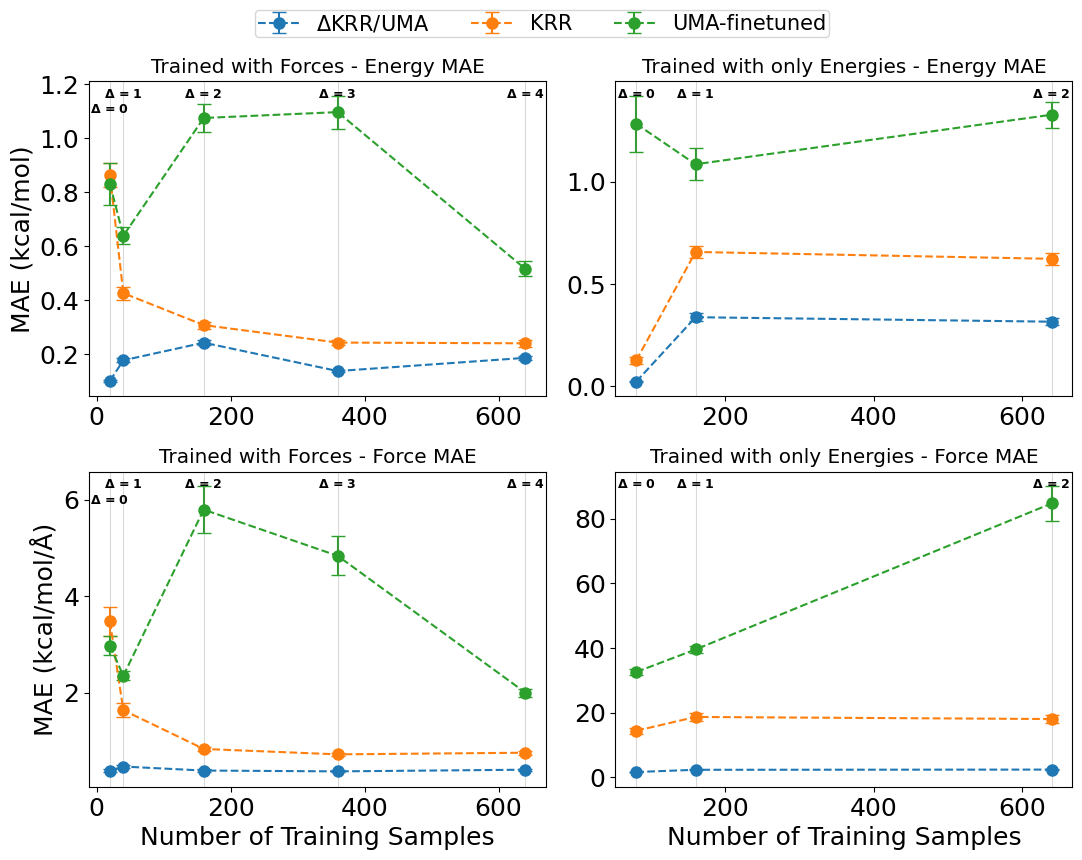
Модели машинного обучения, такие как построенные на основе Kernel Ridge Regression (KRR), устанавливают функциональную зависимость между координатами атомов и энергией системы. Для обучения этих моделей используются данные, полученные в результате высокоточных, но вычислительно затратных, расчетов ab initio, таких как Hartree-Fock или Density Functional Theory (DFT). В процессе обучения, модель KRR аппроксимирует потенциальную энергию системы (E(R), где R — вектор координат атомов) на основе набора тренировочных данных, состоящих из пар (R_i, E_i). Точность полученной модели напрямую зависит от качества и объема тренировочных данных, а также от выбора параметров ядра KRR.
Метод Δ-обучения (дельта-обучения) представляет собой усовершенствование в построении MLFF, заключающееся в моделировании разности между энергиями, рассчитанными с использованием высоко- и низкоуровневых теоретических подходов. Вместо непосредственного предсказания полной энергии системы, модель обучается предсказывать ΔE = E_{high} - E_{low}, где E_{high} — энергия, полученная из более точного, но вычислительно дорогого метода (например, CCSD(T)), а E_{low} — энергия, рассчитанная с использованием менее точного, но быстрого метода (например, DFT). Такой подход значительно повышает переносимость (transferability) модели на различные химические системы и снижает объем требуемых обучающих данных, поскольку разности энергий менее чувствительны к деталям системы, чем абсолютные значения энергии.
Фазово-Лишенный Вспомогательный Монте-Карло: Высокоточный Эталон
Метод фазово-лишенного вспомогательного поля Монте-Карло (ph-AFQMC) представляет собой высокоточный метод Монте-Карло (QMC), предназначенный для решения задач, связанных с сильно коррелированными системами. Он демонстрирует благоприятную масштабируемость по сравнению с другими методами QMC, что позволяет эффективно исследовать системы с большим числом частиц. В отличие от некоторых других QMC методов, ph-AFQMC не требует вычисления фазовых факторов, что упрощает вычисления и снижает вычислительную сложность, сохраняя при этом высокую точность для широкого спектра задач квантовой химии и физики конденсированного состояния.
Метод phaseless Auxiliary-Field Quantum Monte Carlo (ph-AFQMC) использует пробные волновые функции, построенные на основе теории Хартри-Фока. Для минимизации дисперсии и повышения точности расчетов применяются такие методы, как поправка Пулая. Использование пробных функций, основанных на решениях Хартри-Фока, обеспечивает разумный начальный подход к решению многочастичной задачи, а применение поправки Пулая позволяет корректировать энергию, приближая ее к точному значению и снижая статистический шум, возникающий в процессе Монте-Карло симуляции. Данный подход позволяет достичь высокой точности в расчетах для сильно коррелированных систем.
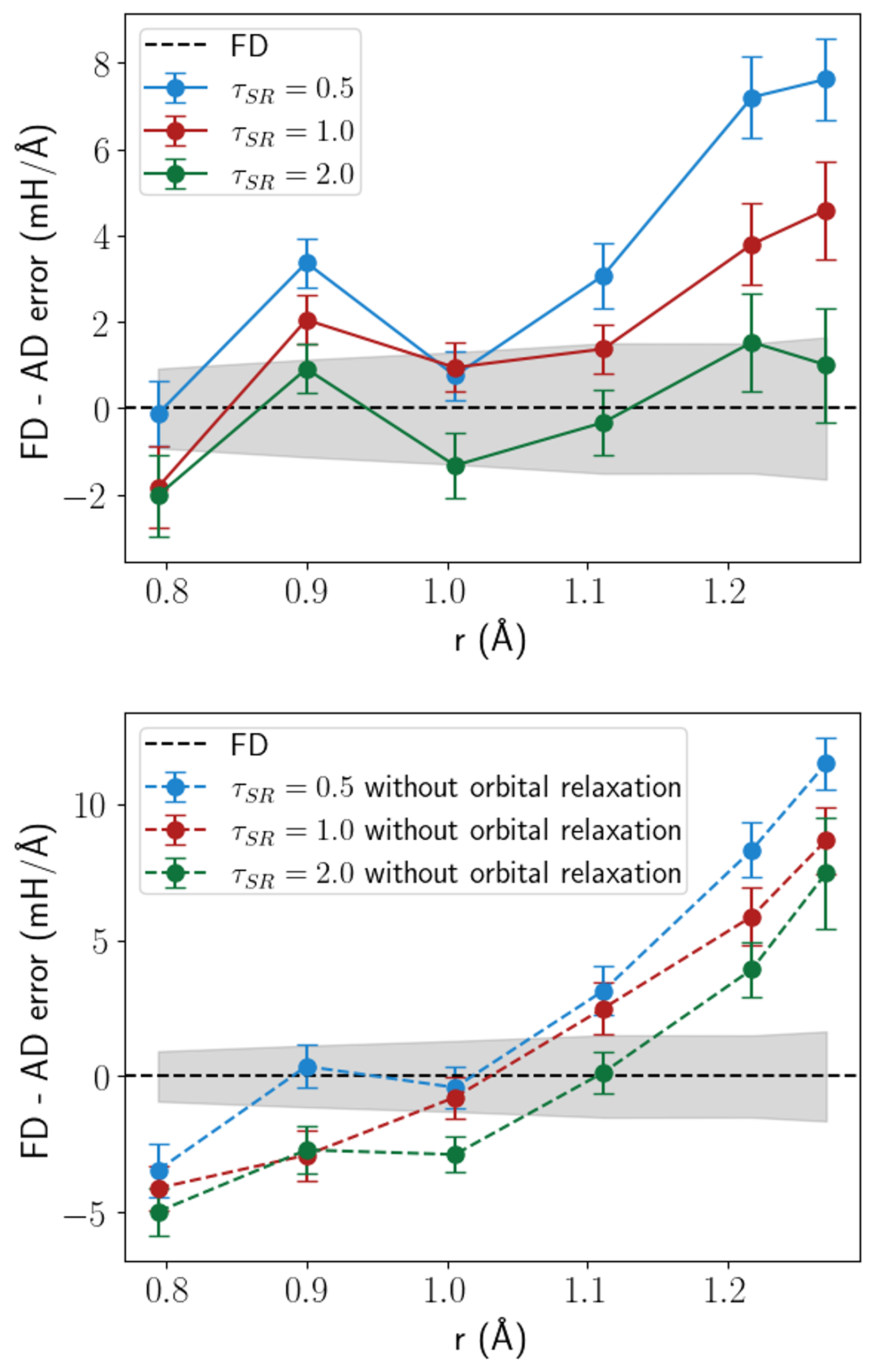
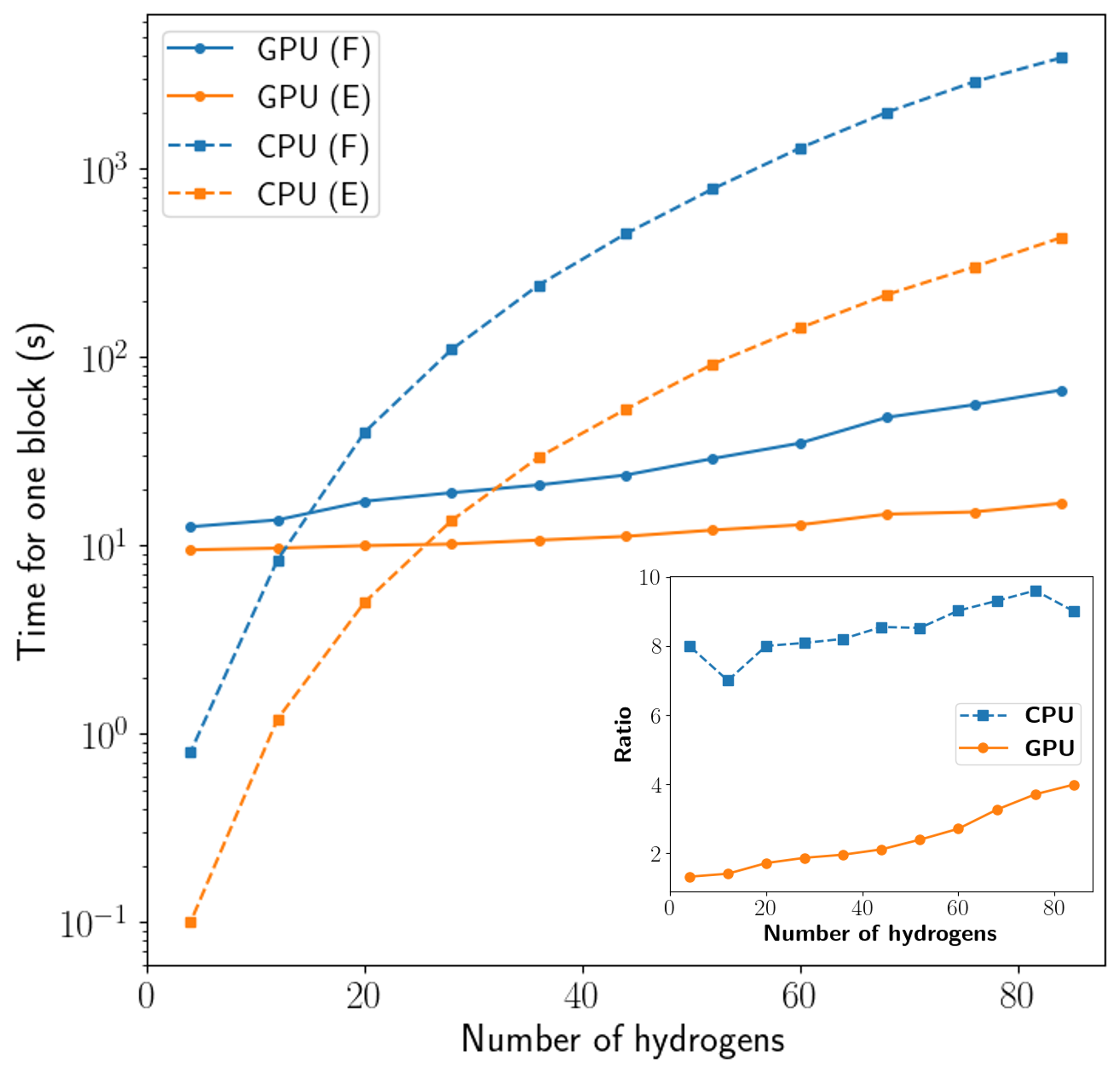
Использование ло́вдиновских ортонормированных атомных орбиталей значительно повышает эффективность и стабильность расчетов в методе ph-AFQMC, предоставляя надежный эталон для валидации моделей машинного обучения межатомных потенциалов (MLFF). В данной работе продемонстрирован метод вычисления градиентов в ph-AFQMC, стоимость которого составляет всего 2-4 раза больше, чем стоимость вычисления энергии на графических процессорах (GPU), и 8-10 раз больше на центральных процессорах (CPU). Это делает вычисление градиентов практически осуществимым для систем, где стоимость вычисления энергии является значительной.
Применение и Валидация: От Переходных Состояний к Молекулярной Динамике
Сочетание фотонного AFQMC (ph-AFQMC) и машинного обучения силовых полей (MLFFs) открывает новые возможности для исследования сложных реакционных путей. Данный подход позволяет эффективно вычислять переходные состояния, критически важные для понимания химических реакций. Метод Nudged Elastic Band (NEB), используемый в сочетании с высокоточными MLFFs, обученными на данных ph-AFQMC, обеспечивает надежный поиск минимальных энергетических точек вдоль реакционной координаты. Благодаря этому, становится возможным детальное изучение механизмов реакций, которые ранее были недоступны для исследования из-за вычислительных ограничений. Такой симбиоз квантово-химических расчетов и методов машинного обучения значительно расширяет границы применимости для моделирования химических процессов.
Молекулярные динамические симуляции, использующие потенциалы, обученные с помощью методов ph-AFQMC, открывают возможности для изучения динамического поведения молекул и материалов в течение продолжительных периодов времени. Такой подход позволяет исследовать сложные процессы, протекающие в системах, от колебаний атомов до крупномасштабных структурных изменений. В отличие от традиционных методов, ограниченных временными масштабами, симуляции, опирающиеся на высокоточные потенциалы, позволяют отслеживать эволюцию систем вплоть до миллисекунд и даже более длительных временных интервалов. Это особенно важно для изучения таких явлений, как свертывание белков, диффузия в твердых телах и химические реакции, протекающие в конденсированной фазе. Благодаря сочетанию точности квантовохимических расчетов и эффективности молекулярной динамики, становится возможным детальное понимание механизмов, определяющих свойства и поведение веществ на атомном уровне.
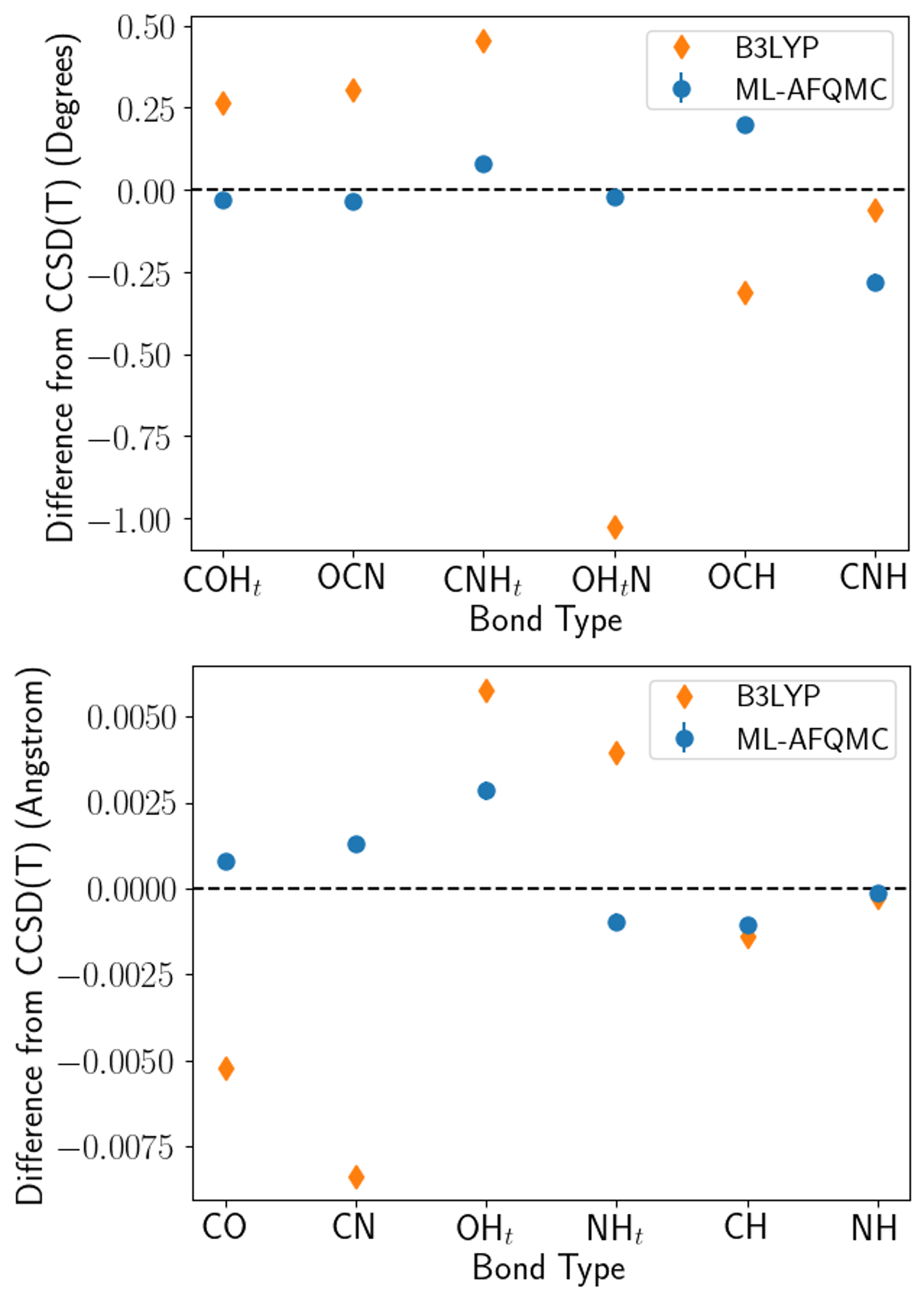
Выполненные расчеты продемонстрировали высокую степень точности, что подтверждается незначительными отклонениями в длинах связей и углах — менее 0.001 Å и 0.1° соответственно. В частности, энергия активации для таутомеризации формамида была определена как 45.23(4) ккал/моль, что находится в полном согласии с результатами, полученными с использованием метода CCSD(T) — общепризнанного «золотого стандарта» в квантовой химии. Достигнутая точность позволяет надежно использовать разработанные методы для моделирования сложных химических процессов и предсказания свойств молекул и материалов с высокой степенью достоверности, открывая новые возможности для разработки инновационных технологий и материалов.
Исследование градиентов ядерных сил, представленное в данной работе, напоминает о сложности взращивания систем, а не построения их. Авторы предлагают метод, сочетающий дифференцирование и машинное обучение, чтобы повысить точность и скорость вычислений в рамках ph-AFQMC. Это не просто оптимизация алгоритма, а скорее создание экосистемы, где каждый выбор архитектуры предсказывает будущие сбои и требует постоянной адаптации. Как говорил Эрнест Резерфорд: «Если бы я мог бы управлять вселенной, я бы не стал». Подобно тому, как невозможно полностью контролировать фундаментальные силы природы, так и в создании сложных вычислительных моделей всегда присутствует элемент непредсказуемости и необходимость учитывать взросление системы.
Что дальше?
Представленный подход к вычислению ядерных градиентов в рамках ph-AFQMC, несомненно, открывает новые возможности для оптимизации геометрии и поиска переходных состояний. Однако, каждый новый инструмент, обещающий упрощение, неминуемо порождает новые сложности. Автоматическое дифференцирование, как и любое другое средство ускорения, лишь переносит бремя вычислительных затрат — теперь уже на этап вычисления производных. Иллюзия контроля над хаосом, создаваемая точными градиентами, быстро развеивается при столкновении с реальными молекулярными системами, где порядок — лишь временный кэш между сбоями.
Настоящим вызовом представляется не столько повышение скорости вычислений, сколько разработка методов, устойчивых к неизбежным ошибкам. Использование машинного обучения, безусловно, перспективно, но требует осторожного подхода. Каждый «обученный» потенциал — это компромисс между точностью и обобщающей способностью. Система, выращенная на ограниченном наборе данных, рискует дать сбой в неожиданной области фазового пространства.
Будущее, вероятно, за гибридными подходами, сочетающими точность квантово-механических расчетов с гибкостью машинного обучения, но при этом осознающими границы применимости каждого из инструментов. Архитектура, обещающая абсолютную свободу, всегда потребует жертвоприношений DevOps — будь то вычислительные ресурсы, время разработчиков или точность результатов. И это не недостаток, а закономерность.
Оригинал статьи: https://arxiv.org/pdf/2602.13187.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Внимание на границе: почему трансформеры нуждаются в «поглотителях»
- Химический синтез под контролем искусственного интеллекта: новые горизонты
- Внимание в сети: Новый подход к ускорению больших языковых моделей
- Искусственный нос будущего: как квантовая механика и машинное обучение распознают запахи
- Физика под контролем: Как «научить» модели понимать мир
- Когда большая языковая модель молчит: как избежать галлюцинаций при ответе на вопросы?
- Пространственная Архитектура для Эффективного Ускорения Нейросетей
- Рассуждения на графах: как большие языковые модели учатся видеть мир
- Гибкие нейросети: как динамическая выборка меняет правила игры
- Грань между Творчеством и Риском: Искусственный Интеллект и Эротический Контент
2026-02-16 23:57