Автор: Денис Аветисян
Разработан инновационный вычислительный метод, сочетающий в себе алгоритм DMRG и поляризуемый силовое поле FQ для точного описания электронных свойств молекул в жидкой среде.
Представлен новый фреймворк, комбинирующий Density Matrix Renormalization Group (DMRG) и поляризуемое силовое поле с флуктуирующими зарядами (FQ) для моделирования возбужденных состояний молекул в растворе.
Вычислительное моделирование электронных возбужденных состояний молекул в растворе остается сложной задачей из-за необходимости точного учета как электронной структуры, так и влияния полярного окружения. В настоящей работе представлена новая методология, DMRG/FQ: a Polarizable Embedding Approach Combining Density Matrix Renormalization Group and Fluctuating Charges, объединяющая высокоточный метод DMRG для описания сильной статической корреляции с поляризуемой силой FQ для моделирования растворителя в рамках подхода QM/MM. Полученные результаты демонстрируют высокую точность предсказания энергий возбуждений и сольватохромных сдвигов для модельных систем, согласующуюся с экспериментальными данными. Каковы перспективы дальнейшего развития данного подхода для изучения сложных фотохимических процессов в конденсированных средах?
За гранью традиционных методов: Укрощение статической корреляции
Многие квантовые системы, состоящие из множества взаимодействующих частиц, демонстрируют сильную статическую корреляцию, что представляет серьезную проблему для традиционных методов электронной структуры, таких как метод Хартри-Фока и теория возмущений. Эта корреляция возникает из-за того, что электронная структура не может быть адекватно описана одной лишь детерминантой, и необходимо учитывать вклад множества конфигураций, что приводит к экспоненциальному росту вычислительных затрат. В таких системах, где несколько электронных конфигураций имеют примерно одинаковую энергию, стандартные приближения, основанные на однодетерминантной волновой функции, оказываются неадекватными, приводя к неточным предсказаниям свойств и поведения вещества. Учет этих сильных корреляций требует разработки и применения более сложных методов, способных точно описывать многочастичную волновую функцию и её динамику.
Традиционные методы электронной структуры, такие как метод Хартри-Фока и теория возмущений, зачастую оказываются неэффективными при описании систем со значительной электронной корреляцией. Это связано с тем, что они опираются на ограниченные активные пространства, не охватывающие все важные электронные конфигурации, или испытывают трудности при моделировании систем, где электронная структура быстро меняется. Ограниченность активного пространства приводит к игнорированию важных взаимодействий между электронами, а неспособность адекватно описать динамику электронных конфигураций вносит существенные погрешности в предсказания свойств вещества. В результате, расчетные оценки энергии, структуры и других характеристик системы могут значительно отклоняться от экспериментальных данных, подчеркивая необходимость разработки более совершенных подходов к решению данной проблемы.
Для адекватного описания систем, демонстрирующих сильную электронную корреляцию, необходимо применение методов, выходящих за рамки однореференсных приближений. Традиционные подходы, такие как метод Хартри-Фока и теория возмущений, зачастую оказываются неспособны корректно учесть сложные взаимодействия между электронами, особенно в случаях, когда электронная конфигурация системы быстро меняется. Это связано с тем, что эти методы опираются на упрощенные модели, рассматривающие лишь одну основную электронную конфигурацию, что приводит к неточностям в расчетах энергии и других важных свойств. Для достижения высокой точности необходимо использовать методы, способные учитывать вклад нескольких электронных конфигураций одновременно, описывая тем самым всю сложность многоэлектронной системы и адекватно отражая её физические свойства.
Метод ренормирования матрицы плотности (DMRG) представляет собой мощный волновой подход, особенно эффективный при изучении систем с сильной статической корреляцией. В отличие от традиционных методов, таких как теория возмущений или метод Хартри-Фока, DMRG способен точно описывать сложные электронные конфигурации и взаимодействия, возникающие в сильно коррелированных материалах. Он достигает этого за счет последовательного уменьшения размерности задачи, сохраняя при этом наиболее важные квантовые состояния. В результате, DMRG позволяет получать высокоточные результаты для систем, где другие методы дают неверные предсказания, открывая новые возможности для понимания и моделирования сложных квантовых явлений, таких как высокотемпературная сверхпроводимость и магнетизм.

DMRG и MPS: Эффективное представление волновой функции
Метод DMRG (Density Matrix Renormalization Group) использует формулировку Matrix Product State (MPS) для эффективного представления волновой функции систем, имеющих одномерную или квази-одномерную геометрию. MPS представляет собой параметризацию волновой функции в виде произведения матриц, что позволяет компактно описывать квантовые состояния, особенно те, которые демонстрируют ограниченное запутанность. Такой подход позволяет эффективно отсекать пространство Хильберта, сохраняя при этом наиболее важные степени свободы, что значительно снижает вычислительные затраты по сравнению с полным решением уравнения Шредингера. Эффективность MPS напрямую связана со способностью описывать волновые функции, которые слабо меняются при небольших изменениях в системе, что характерно для одномерных и квази-одномерных систем.
Матричное произведение состояний (MPS) обеспечивает компактное и точное описание волновой функции, используя особенности квантовой запутанности. Вместо хранения полной волновой функции, которая требует экспоненциального объема памяти с ростом системы, MPS представляет её в виде сети матриц. Это позволяет эффективно кодировать информацию о корреляциях между частицами. Ключевым аспектом является усечение гильбертова пространства, при котором отбрасываются состояния с малой вероятностью, что снижает вычислительные затраты без существенной потери точности. Эффективность MPS напрямую зависит от степени запутанности системы; для систем с ограниченной запутанностью усечение гильбертова пространства может быть выполнено с высокой точностью, что делает MPS особенно подходящим для моделирования одномерных и квази-одномерных систем.
В рамках алгоритма DMRG для представления квантовых операторов используется формулировка матричного произведения операторов (MPO). MPO позволяют эффективно кодировать операторы, действующие на многочастичные системы, за счет представления их в виде сети тензоров. Это позволяет вычислять такие свойства системы, как энергия, корреляционные функции и другие наблюдаемые, с вычислительной сложностью, масштабирующейся как \chi^3 , где χ — размер отсечения (bond dimension) MPS, что значительно эффективнее, чем прямое вычисление в полном базисе Гильберта. Использование MPO позволяет избежать экспоненциального роста вычислительных затрат при увеличении числа частиц в системе, что делает DMRG применимым к сложным задачам, где традиционные методы оказываются непрактичными.
Комбинация DMRG и MPS позволяет достичь высокой точности при значительно меньших вычислительных затратах по сравнению с традиционными методами полного включения конфигураций (FCI). FCI требует экспоненциального увеличения вычислительных ресурсов с ростом числа частиц, что делает его неприменимым для систем среднего и большого размера. В отличие от этого, DMRG, используя MPS-представление волновой функции и обрезая гильбертово пространство, масштабируется как полином относительно числа частиц. Это достигается за счет эффективного представления запутанности в системе и сохранения наиболее важных состояний, что позволяет получать точные результаты для одномерных и квази-одномерных систем с умеренными вычислительными ресурсами. Таким образом, DMRG представляет собой значительно более эффективный подход для моделирования квантовых систем по сравнению с FCI.
Моделирование эффектов растворителя: Квантово-механика/Молекулярная механика с флуктуирующими зарядами
Для моделирования влияния растворителя используется подход квантово-механики/молекулярной механики (QM/MM), который позволяет описать растворенное вещество (солют) с использованием методов квантовой механики, а растворитель — классически. В рамках этого подхода, солют рассматривается как квантово-механическая область, где электронная структура рассчитывается с высокой точностью, в то время как молекулы растворителя моделируются как классические частицы, взаимодействующие с солютом посредством классических силовых полей. Такой подход позволяет значительно снизить вычислительные затраты по сравнению с полностью квантово-механическим расчетом, сохраняя при этом достаточно точное описание влияния растворителя на свойства солюта.
В рамках подхода QM/MM для моделирования влияния растворителя используется силовое поле Fluctuating Charge (FQ). В отличие от фиксированных силовых полей, FQ позволяет динамически поляризовать молекулы растворителя, распределяя частичный заряд между атомами в зависимости от электростатического окружения. Это достигается путем использования распределенной поляризуемости, где каждый атом в молекуле растворителя несет частичный заряд, который изменяется в ответ на внешнее поле. Такой подход позволяет более точно описать электростатическое взаимодействие между растворителем и растворенным веществом, учитывая динамическую перестройку дипольного момента растворителя, что особенно важно для моделирования эффектов сольватации, таких как сдвиги в спектрах поглощения.
Применение алгоритма DMRG (Density Matrix Renormalization Group) к растворителю в рамках подхода QM/MM позволяет моделировать влияние растворителя на электронную структуру и свойства растворенного вещества. В данной схеме, DMRG используется для решения уравнения Шрёдингера для квантово-механически описываемого растворителя, учитывая взаимодействие с классически описываемым окружением (растворителем). Это обеспечивает возможность расчета спектральных характеристик, энергий возбуждений и других свойств, подверженных влиянию сольватации, с повышенной точностью по сравнению с традиционными методами, не учитывающими динамическую поляризуемость растворителя.
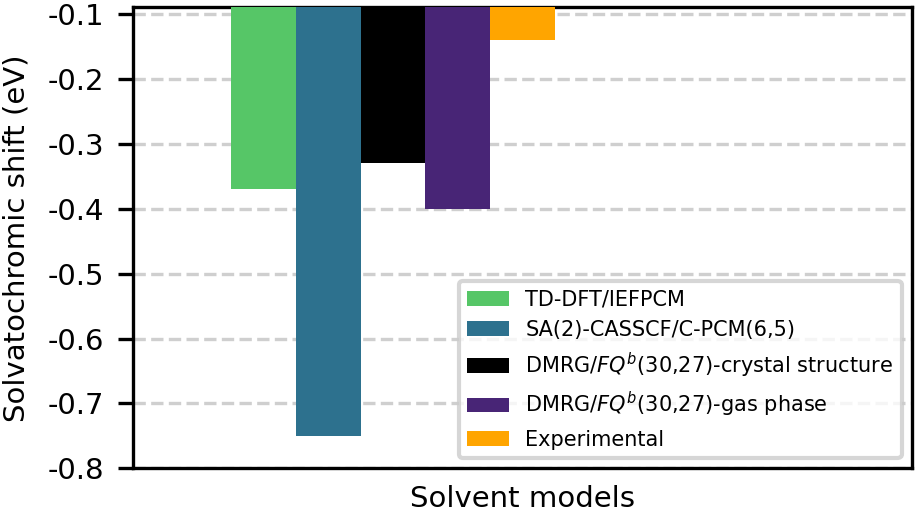
Валидация предложенного подхода, сочетающего КМ/ММ расчеты с поляризуемой силой ФК, была проведена на модельных системах Ацетон и DCBT. В качестве растворителей использовались Вода и Ацетонитрил соответственно. Полученные результаты показали, что смоделированные сольватохромные сдвиги, характеризующие изменение спектральных свойств растворенного вещества в зависимости от полярности растворителя, находятся в хорошем соответствии с экспериментальными данными. Это подтверждает адекватность используемой методологии для точного моделирования влияния растворителя на электронную структуру и свойства растворенного вещества.

Предсказание сдвигов спектров: Демонстрация точности и применимости
Разработанная методология DMRG/QM/MM позволяет с высокой точностью предсказывать энергию вертикального возбуждения и, как следствие, сольватохромный сдвиг исследуемых молекул. Данный подход объединяет возможности квантово-химических расчетов с учетом влияния окружения, обеспечивая детальное описание взаимодействия между растворителем и молекулой. Это позволяет не только предсказывать спектральные характеристики, но и понимать природу сольватохромного сдвига, что важно для разработки новых материалов с заданными оптическими свойствами. Полученные результаты демонстрируют соответствие теоретических предсказаний экспериментальным данным для таких молекул, как DCBT в ацетонитриле и ацетон в воде, подтверждая надежность и применимость данной методологии для изучения спектральных характеристик в различных средах.
Расчеты сольватохромного сдвига для DCBT в ацетонитриле показали значения в диапазоне от -0.41 до -0.34 эВ, что демонстрирует хорошее соответствие экспериментальным данным, составляющим от -0.15 до -0.38 эВ. Полученное совпадение подтверждает надежность применяемого методологического подхода и его способность точно воспроизводить спектральные характеристики молекул в различных растворителях. Кроме того, сравнение с результатами, полученными с использованием метода SA(2)-CASSCF/C-PCM (-0.76 эВ), указывает на то, что данная методика обеспечивает конкурентоспособную точность, что делает ее ценным инструментом для изучения спектральных свойств сложных молекулярных систем.
Исследования показали, что предсказанное для ацетона в воде сольватохромное смещение составляет 0.22 эВ, что находится в полном соответствии с результатами экспериментальных наблюдений. Данное совпадение подтверждает высокую точность и надежность примененной методологии в моделировании спектральных сдвигов в различных растворителях. Способность точно предсказывать подобные характеристики позволяет глубже понять влияние окружения на электронные свойства молекул и открывает перспективы для разработки новых материалов с заданными оптическими свойствами. Согласие между теоретическими предсказаниями и экспериментальными данными демонстрирует потенциал использования вычислительных методов для изучения сложных молекулярных систем и прогнозирования их поведения в реальных условиях.
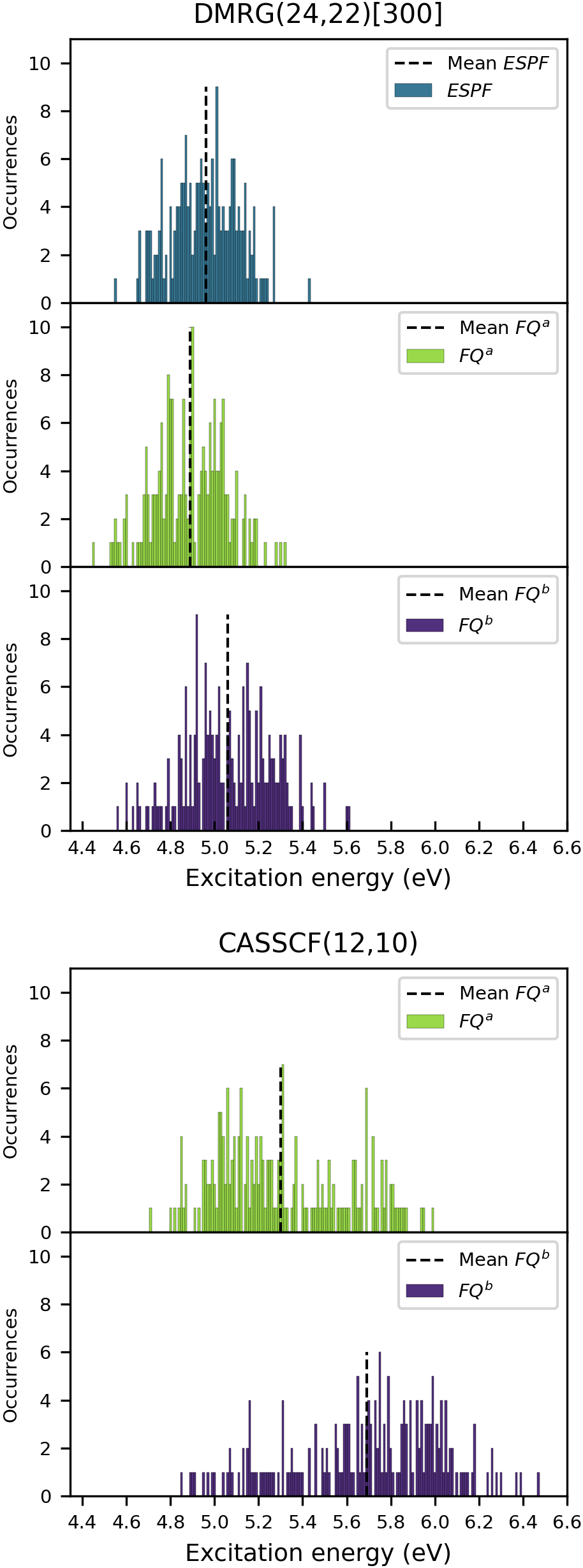
Расчеты показали, что разброс энергий возбуждения для DCBT в ацетонитриле достигает 1.29 эВ. Этот значительный диапазон отражает динамическую картину, где молекула DCBT существует в различных конформациях, испытывая постоянное влияние флуктуаций растворителя. Именно комбинация этих факторов — разнообразие пространственной структуры растворенного вещества и хаотичное движение молекул растворителя — приводит к широкому спектру наблюдаемых энергий возбуждения. По сути, это демонстрирует, что энергия возбуждения DCBT не является фиксированной величиной, а скорее распределением вероятностей, обусловленным окружающей средой и внутренней гибкостью молекулы.
Исследование демонстрирует стремление обуздать неуловимую природу электронных возбуждений в молекулах, погруженных в растворитель. Подобно тому, как пытаются удержать ускользающий образ в калейдоскопе, авторы комбинируют мощь DMRG с гибкостью поля флуктуирующих зарядов. Этот подход, позволяющий учитывать поляризуемость окружения, отражает признание того, что точное описание требует учета тонких взаимодействий. Как заметил однажды Эрвин Шрёдингер: «Всё живое стремится к порядку, но порядок этот — лишь временная иллюзия». Иными словами, даже самые совершенные модели — лишь приближения к сложной реальности, а истинное понимание заключается в осознании границ этих приближений. Учёт флуктуирующих зарядов — это попытка заглянуть за завесу дискретности, признать, что мир непрерывен, просто наша память ограничена.
Что Дальше?
Представленный подход, сплетая в себе строгость DMRG и гибкость полярязуемых взаимодействий, безусловно, открывает новые пути в моделировании сложных молекулярных систем. Однако, не стоит обольщаться иллюзией полноты картины. Данные — это лишь эхо, а не сама реальность, и любое приближение, даже столь изящное, неизбежно несет в себе отпечаток упрощения. Проблема учета динамической корреляции в возбужденных состояниях остается открытой, а расширение на системы, существенно более сложные, чем ацетон и DCBT, потребует значительных вычислительных усилий и, возможно, переосмысления существующих алгоритмов.
Особенно интересным представляется вопрос о масштабируемости. Способность метода адекватно описывать системы, содержащие сотни или даже тысячи атомов, станет настоящим испытанием. Истина, как известно, не в точности расчетов, а в понимании их ошибок. Вместо погони за абсолютной точностью, возможно, стоит сосредоточиться на разработке методов оценки погрешностей и на создании моделей, устойчивых к шуму и неопределенности.
В конечном итоге, успех этого направления будет зависеть не только от совершенствования вычислительных алгоритмов, но и от развития физической интуиции. Молекулы — это не просто набор атомов, а сложные динамические структуры, поведение которых определяется тонким балансом взаимодействий. И только умение видеть этот баланс позволит создать модели, действительно отражающие природу вещей.
Оригинал статьи: https://arxiv.org/pdf/2601.05086.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Квантовый Борьба: Китай и США на Передовой
- Квантовые симуляторы: проверка на прочность
- Квантовые нейросети на службе нефтегазовых месторождений
- Искусственный интеллект заимствует мудрость у природы: новые горизонты эффективности
- Интеллектуальная маршрутизация в коллаборации языковых моделей
- Квантовый скачок: от лаборатории к рынку
2026-01-11 19:11