Автор: Денис Аветисян
Исследование демонстрирует, что алгоритмы, имитирующие квантовые процессы, предлагают эффективный и масштабируемый подход к вычислению энергии основного состояния молекул.
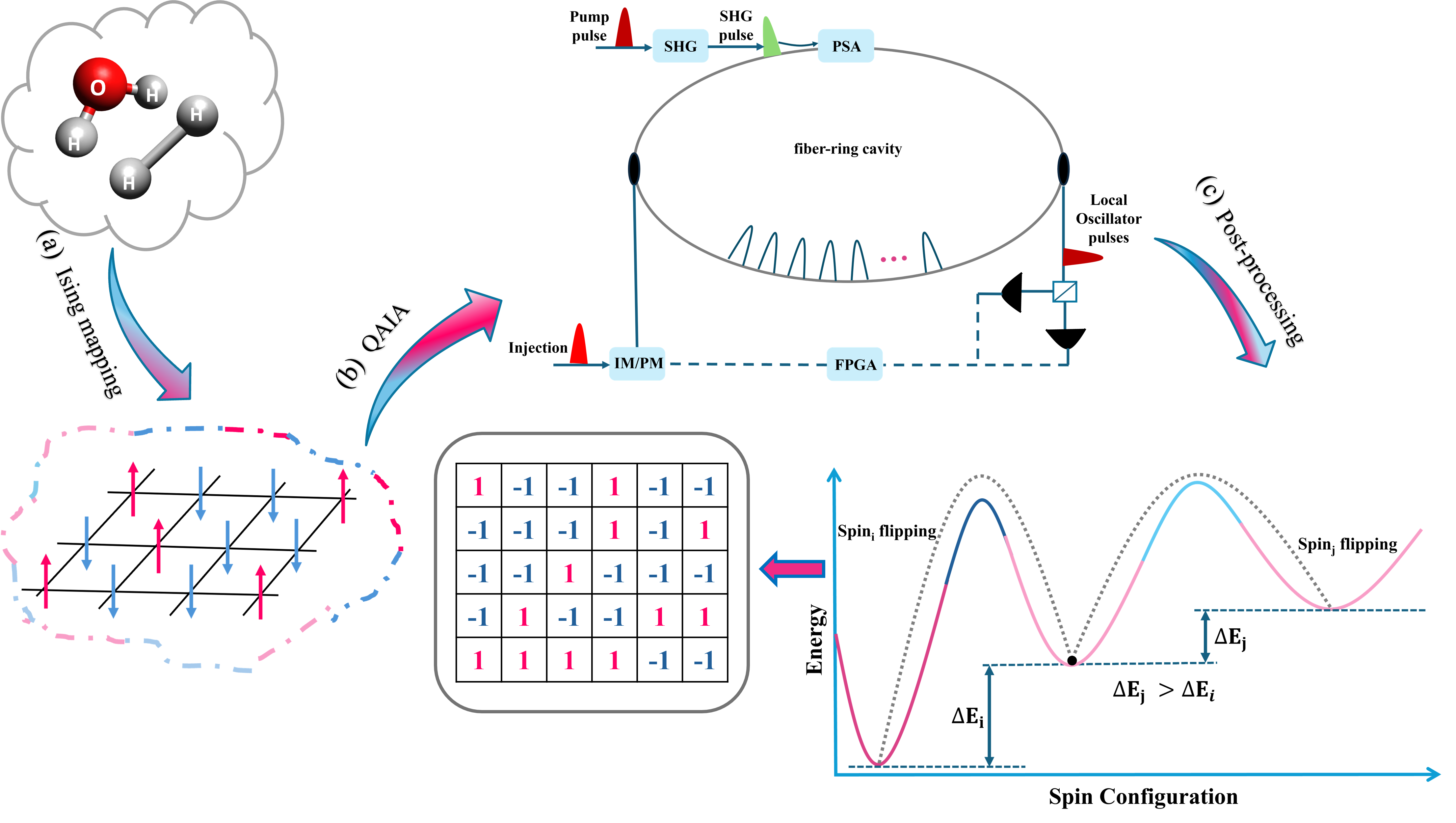
В статье рассматриваются возможности Когерентных Машин Изинга и Симулированного Разветвления для решения задач квантовой химии с использованием GPU-ускорения.
Несмотря на значительный прогресс в квантовых вычислениях, шум в современных устройствах остается серьезным препятствием для моделирования сложных молекулярных систем. В работе ‘Quantum-Inspired Ising Machines for Quantum Chemistry Calculations’ представлено исследование, демонстрирующее, что квантово-вдохновленные алгоритмы, в частности когерентные машины Изинга и алгоритмы моделирования бифуркаций, способны эффективно вычислять профили электронной энергии молекул H₂ и H₂O. Полученные результаты свидетельствуют о существенном ускорении по сравнению с традиционными подходами на основе квантовых вентильных схем, открывая перспективы для масштабирования вычислений на более крупные системы. Смогут ли эти методы стать основой для новых подходов в химии и материаловедении?
Вызов Точной Молекулярной Симуляции
Вычислительные методы, стремящиеся к максимальной точности при описании электронного строения молекул, такие как полная конфигурационная интеграция (FCI), сталкиваются с серьезными ограничениями. Несмотря на теоретическую способность предоставить точное решение $Schrödinger$ уравнения, вычислительные затраты растут экспоненциально с увеличением числа электронов и атомных орбиталей. Это означает, что применение FCI становится практически невозможным даже для относительно небольших молекул, состоящих из нескольких атомов. Фактически, сложность вычислений ограничивает возможность исследования сложных химических систем и материалов, что подчеркивает необходимость разработки более эффективных и приближенных методов.
Традиционные методы расчета электронной структуры, такие как метод Хартри-Фока и Complete Active Space Configuration Interaction (CASCI), обеспечивают значительное ускорение вычислений, однако достигается это за счет внесения упрощающих приближений в исходный молекулярный гамильтониан $Ĥ$. Эти приближения, как правило, связаны с пренебрежением корреляцией электронов, что приводит к неточному описанию энергетических уровней и свойств молекул. Хотя CASCI и более точно учитывает корреляцию, чем метод Хартри-Фока, его применение ограничено небольшими активными пространствами из-за экспоненциального роста вычислительных затрат с увеличением числа электронов и базисных функций. В результате, несмотря на свою скорость, эти методы могут давать существенные погрешности при моделировании сложных химических систем и прогнозировании их поведения.
Ограничение, возникающее из-за компромисса между точностью и скоростью вычислений, существенно препятствует моделированию сложных химических систем и разработке инновационных материалов. В частности, для молекул, содержащих большое количество электронов и атомов, точное решение $Шрёдингеровского$ уравнения становится непосильной задачей для современных вычислительных мощностей. Это означает, что предсказание свойств, таких как реакционная способность, спектральные характеристики или структура новых соединений, требует упрощений, которые могут привести к значительным ошибкам. В результате, создание материалов с заданными свойствами, оптимизация каталитических процессов или понимание сложных биологических механизмов, зависящих от точного описания электронного строения, сталкивается с серьезными трудностями, требующими разработки новых, более эффективных вычислительных методов.
Приручение Квантовой Мощи: Гибридные Алгоритмы Вступают в Игру
Вариационный квантовый эйнзольвер (VQE) представляет собой гибридный квантово-классический алгоритм, предназначенный для нахождения основного состояния энергии квантовых систем. Алгоритм сочетает в себе использование квантового компьютера для оценки энергии предложенной волновой функции и классического оптимизатора для итеративной минимизации этой энергии. VQE особенно перспективен для применения на устройствах NISQ (Noisy Intermediate-Scale Quantum), поскольку он разработан с учетом ограниченных возможностей и подверженности ошибкам этих устройств. В отличие от полностью квантовых алгоритмов, VQE снижает требования к когерентности кубитов и глубине квантовых цепей, делая его более реалистичным для текущего поколения квантового оборудования. Основной целью является приближенное решение уравнения Шрёдингера $H|\psi\rangle = E|\psi\rangle$, где $H$ — гамильтониан системы, $E$ — энергия, а $|\psi\rangle$ — основное состояние.
Эффективность алгоритма VQE напрямую зависит от выбора анзаца — пробной волновой функции, используемой для аппроксимации истинного основного состояния. Два широко используемых варианта — UCCSD (Unitary Coupled Cluster with Single and Double excitations) и k-UpCCGSD (k-Up Coupled Cluster with Single, Double, and potentially higher excitations). UCCSD обеспечивает относительно высокую точность, но требует значительных квантовых ресурсов. k-UpCCGSD представляет собой обобщение UCCSD, позволяющее контролировать уровень усечения кластеров возбуждений, что дает возможность балансировать между точностью и сложностью вычислений. Выбор конкретного анзаца определяется сложностью решаемой задачи и доступными ресурсами квантового оборудования.
Методы ADAPT-VQE и Qubit-ADAPT-VQE представляют собой адаптивные алгоритмы, динамически изменяющие структуру пробной волновой функции (анзаца) в процессе оптимизации. В отличие от фиксированных анзацев, таких как UCCSD, эти подходы добавляют или удаляют квантовые гейты на основе информации, полученной в ходе вычислений. Qubit-ADAPT-VQE, в частности, оптимизирует анзац на уровне отдельных кубитов, что позволяет более эффективно адаптировать его к конкретной задаче и потенциально снизить требования к количеству кубитов и глубине квантовой цепи, необходимых для достижения заданной точности при расчете основного состояния энергии. Такой подход направлен на повышение эффективности Variational Quantum Eigensolver (VQE) на устройствах NISQ.
Квантовый Отжиг и Его Классические Отражения
Квантовый отжиг (QA) использует квантовые флуктуации для поиска состояния с минимальной энергией системы, описываемой гамильтонианом Изинга. Гамильтониан Изинга представляет собой математическую модель, используемую в статистической механике для описания ферромагнетиков и других систем, состоящих из взаимодействующих спинов. В QA, система эволюционирует от простого начального состояния к состоянию с минимальной энергией, используя квантовое туннелирование для преодоления энергетических барьеров, что позволяет находить решения оптимизационных задач, которые сложно решить классическими методами. Эффективность QA зависит от способности поддерживать квантовую когерентность на протяжении всего процесса отжига и от точности отображения решаемой задачи на гамильтониан Изинга.
Гибридные квантово-классические алгоритмы, такие как Quantum Annealer Eigensolver (QAE), применяют квантовый отжиг для расчётов в области электронной структуры. В рамках QAE, задача нахождения основного состояния молекулярной системы преобразуется в задачу квадратичной безусловной двоичной оптимизации (QUBO). Это преобразование позволяет использовать квантовый отжиг для поиска минимума энергии, соответствующего основному состоянию. В частности, гамильтониан молекулы отображается на гамильтониан Изинга, который затем формулируется как задача QUBO, пригодная для решения на кванновом отжиговом процессоре. Полученные результаты используются для приближённого вычисления энергии основного состояния и других свойств молекулы.
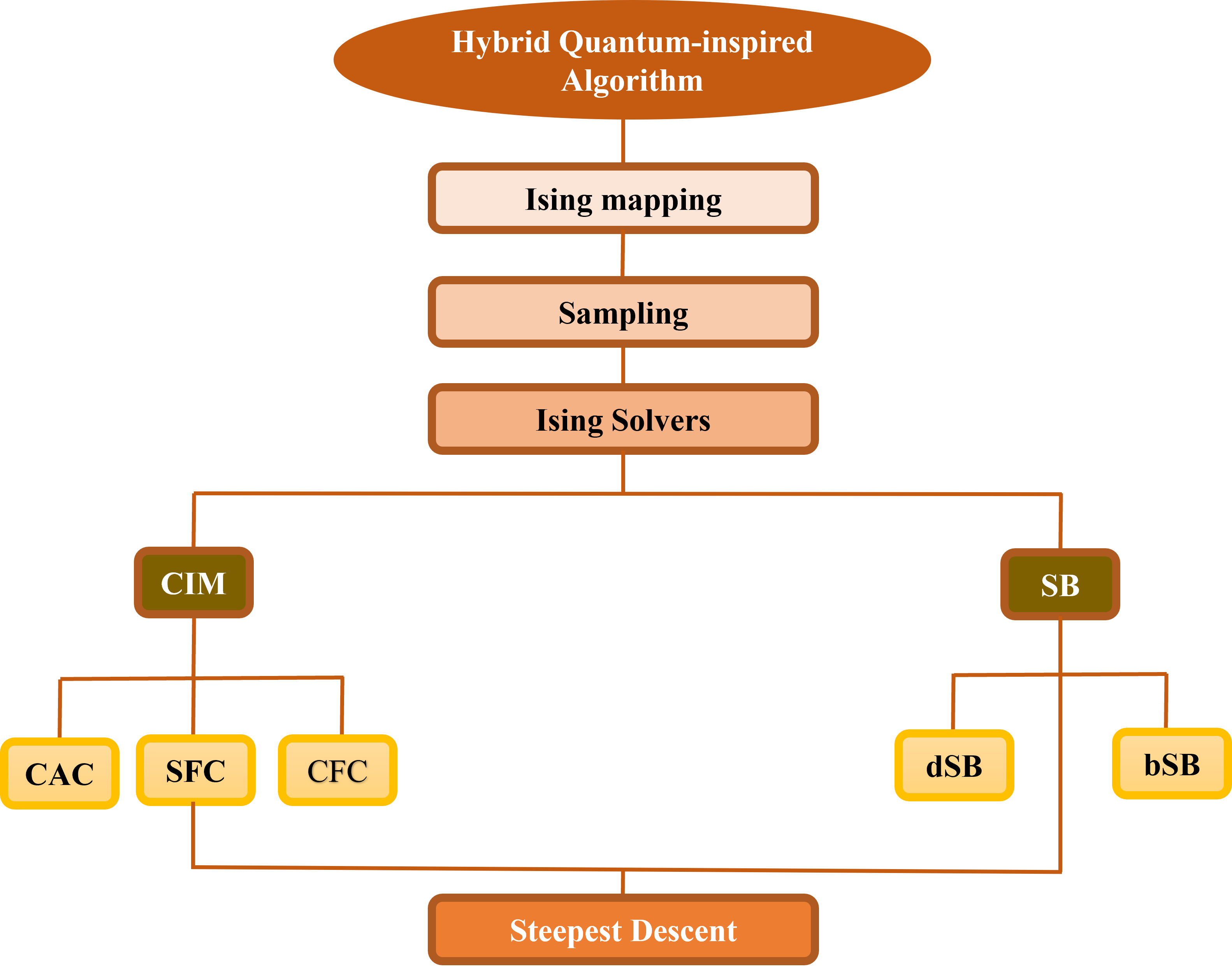
Классические алгоритмы, вдохновленные квантовым отжигом, представляют собой альтернативный подход к задачам оптимизации, реализуемый на традиционном оборудовании. Эти алгоритмы, такие как Quantum Annealing-Inspired Algorithms, имитируют принципы работы квантового отжига, используя методы, такие как Когерентные машины Изинга и Симулированная бифуркация. Когерентные машины Изинга представляют собой аналоговые вычислительные системы, предназначенные для решения задач оптимизации, основанных на модели Изинга, а симулированная бифуркация использует динамику нелинейных систем для поиска оптимальных решений. Оба подхода стремятся к достижению аналогичных преимуществ в оптимизации, что и квантовый отжиг, но без необходимости использования квантового оборудования, что делает их привлекательными для широкого спектра приложений.
Соединяя Разрозненное: Фермионное Отображение и Расширения Алгоритмов
Для успешного применения квантовых алгоритмов к моделированию молекулярных систем необходимо преобразование фермионной задачи в представление, пригодное для кубитов. Этот процесс осуществляется посредством специальных преобразований, среди которых наиболее распространены преобразование Жордана-Вигнера и преобразование Бравьи-Китайева. Данные преобразования позволяют сопоставить фермионные операторы, описывающие электроны в молекуле, с операторами, действующими на кубиты. Преобразование Жордана-Вигнера, хотя и простое в реализации, может приводить к нелокальным операторам, что затрудняет вычисления. Преобразование Бравьи-Китайева, в свою очередь, стремится минимизировать нелокальность, что потенциально ускоряет вычисления, однако оно более сложное в реализации. Выбор оптимального преобразования зависит от конкретной молекулярной системы и доступных вычислительных ресурсов, поскольку оба метода играют ключевую роль в эффективности квантового моделирования.
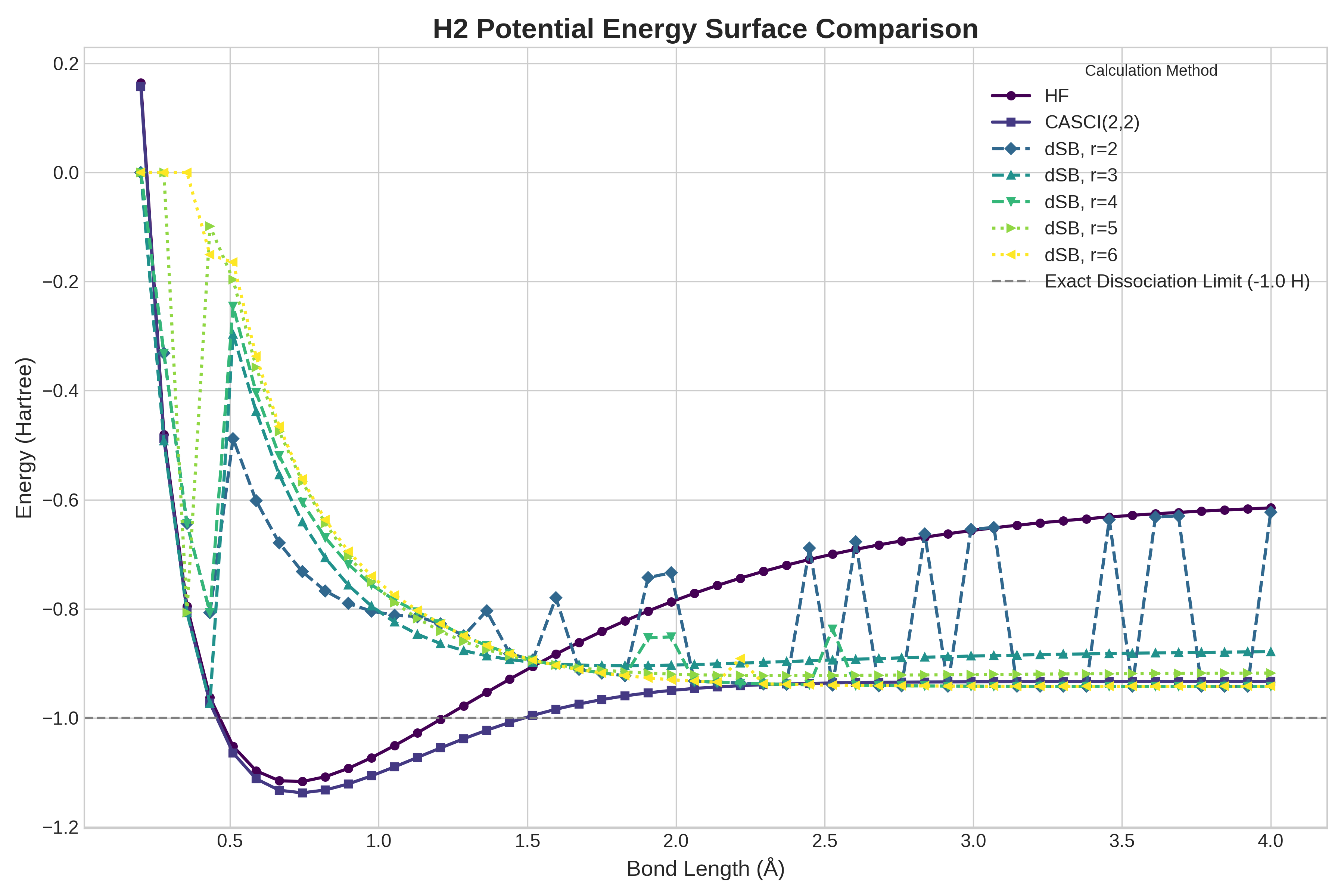
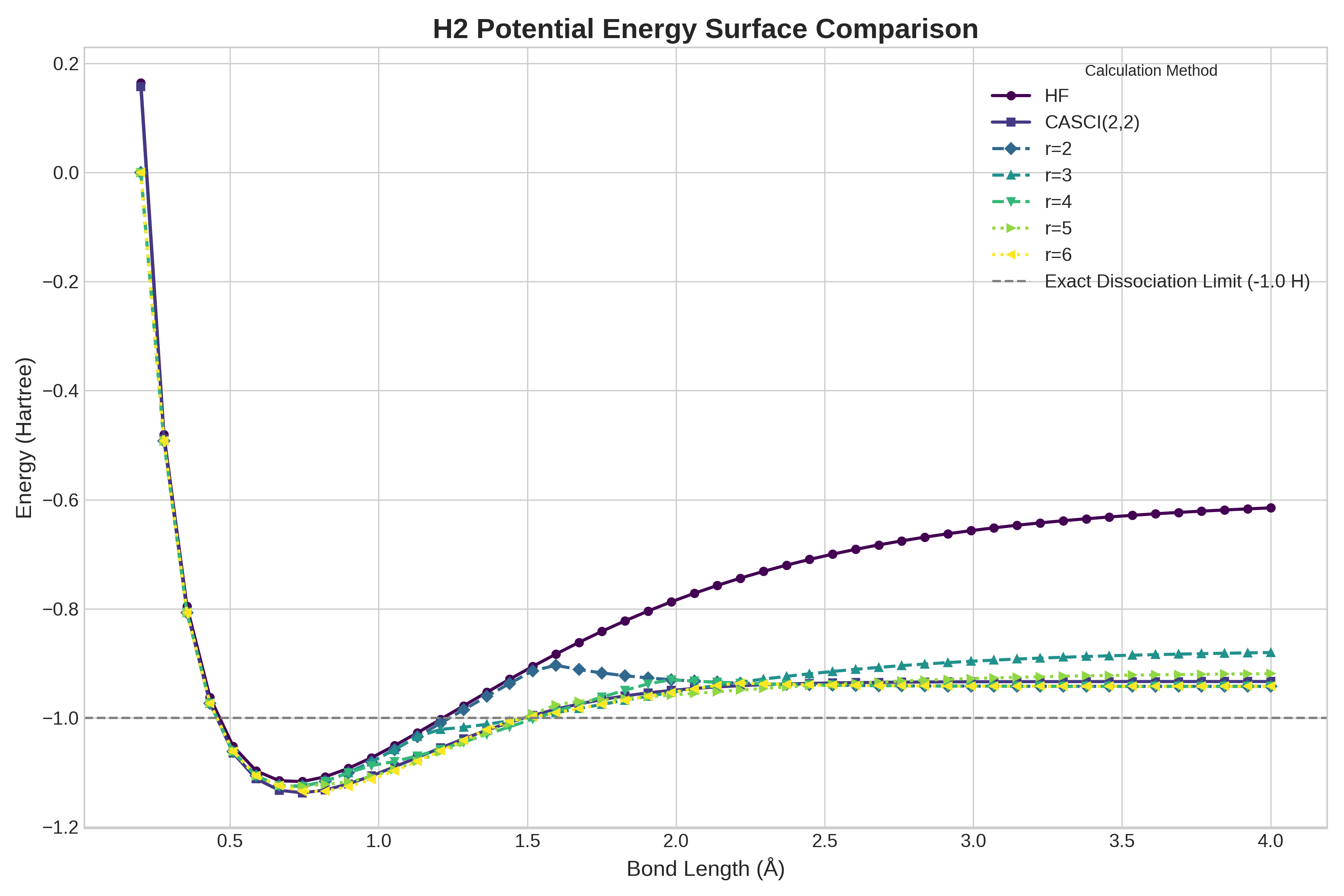
Алгоритмические расширения, такие как dSB (Direct Second-order Belief Propagation) и SFC (Sparse Frequency Complexity), представляют собой важные усовершенствования базовых методов преобразования фермионных проблем в кубитные. Эти подходы направлены на оптимизацию процесса вычислений в квантовой химии, решая специфические задачи, возникающие при моделировании электронных структур молекул. dSB, например, улучшает сходимость и точность путем более эффективного распространения информации о корреляциях между электронами. SFC же фокусируется на снижении вычислительной сложности за счет использования разреженных представлений волновых функций. В совокупности, эти расширения позволяют более эффективно использовать возможности квантовых алгоритмов для решения сложных задач в химии и материаловедении, значительно ускоряя расчеты и повышая точность получаемых результатов, особенно для крупных молекулярных систем.
Исследование продемонстрировало, что разработанный вариант алгоритма Хаотической Обратной Связи (CFC) способен вычислять полные профили энергии для молекул водорода ($H_2$) и воды ($H_2O$) за 1,2 и 2,4 секунды соответственно. При этом точность полученных результатов сопоставима со стандартными квантовыми подходами. Отдельные вычисления с использованием CFC занимают от 1 до 3 секунд на графических процессорах (GPU), что значительно ускоряет процесс моделирования электронных структур и открывает перспективы для исследования более сложных молекулярных систем с высокой скоростью и эффективностью.
Исследования показали, что алгоритм Хаотической Обратной Связи (CFC) значительно превосходит существующее квантовое оборудование по скорости вычисления энергии молекулярных систем. В частности, время оценки энергии для молекулы $H_2$ составляет 334 ± 40 секунд при использовании квантового процессора IBMQ Manila, и 13297 ± 625 секунд на AQT Marmot. Данные результаты демонстрируют существенное ускорение вычислений, предоставляя возможность более эффективного моделирования сложных молекулярных структур и процессов, что открывает новые перспективы в области квантовой химии и материаловедения. Полученное преимущество в скорости вычислений делает алгоритм CFC перспективным инструментом для решения задач, недоступных для традиционных квантовых подходов из-за ограничений аппаратных ресурсов.
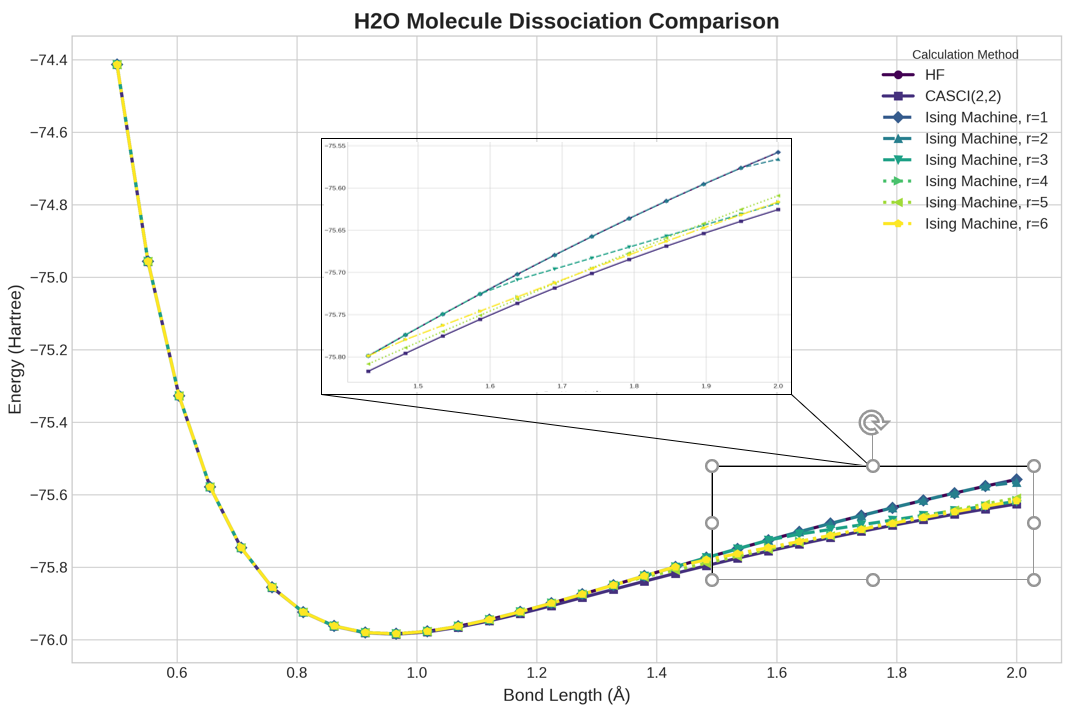
Исследование демонстрирует, что квантово-вдохновленные алгоритмы, такие как машины Коэрентных спинов и моделирование бифуркаций, представляют собой жизнеспособную альтернативу традиционным и квантовым методам вычисления энергии основного состояния молекул. Подобный подход, с акцентом на масштабируемость и эффективность, особенно при использовании GPU-ускорения, неизбежно порождает сложные взаимосвязи, требующие тонкой настройки. Как однажды заметил Джон Белл: «Если бы вы не знали квантовой механики, то думали бы, что это магия». Эта фраза точно отражает суть представленной работы, где сложные вычисления энергии молекул, осуществляемые с помощью вдохновленных квантовыми принципами алгоритмов, кажутся выходящими за рамки привычных представлений о вычислительных возможностях. В контексте исследования, каждый архитектурный выбор, определяющий структуру алгоритма, предсказывает потенциальную уязвимость и необходимость адаптации к постоянно меняющимся условиям.
Что Дальше?
Представленная работа демонстрирует применимость алгоритмов, вдохновленных квантовой механикой — в частности, когерентных машин Изинга и моделирования бифуркаций — для вычисления энергии основного состояния молекул. Однако, оптимизация — это всегда лишь отсрочка неизбежного. Вместо поиска «идеального» алгоритма, следует признать, что каждая архитектурная оптимизация — это пророчество о будущей точке отказа. Ускорение на GPU лишь временно маскирует фундаментальную зависимость системы от аппаратных ограничений.
Разделение задачи на более мелкие компоненты, как это делается в гибридных подходах, не отменяет общей судьбы. Разделение системы — это иллюзия контроля. Всё связанное рано или поздно рухнет синхронно. Будущие исследования должны сосредоточиться не на увеличении масштабируемости, а на понимании закономерностей этих отказов, на разработке механизмов самовосстановления и адаптации, а не на создании всё более сложных и хрупких конструкций.
Эра «решения» задач заканчивается. Наступает эпоха управления сложностью. Вопрос не в том, как найти «лучшее» приближение, а в том, как научиться жить с неизбежной неопределенностью, с фундаментальным ограничением вычислительных ресурсов, и с тем фактом, что любая система стремится к зависимости.
Оригинал статьи: https://arxiv.org/pdf/2512.16435.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Квантовый Борьба: Китай и США на Передовой
- Квантовый скачок: от лаборатории к рынку
- Квантовые симуляторы: проверка на прочность
- Квантовые нейросети на службе нефтегазовых месторождений
- Искусственный интеллект заимствует мудрость у природы: новые горизонты эффективности
- Интеллектуальная маршрутизация в коллаборации языковых моделей
2025-12-19 10:48