Автор: Денис Аветисян
Новый подход на базе искусственного интеллекта позволяет генерировать молекулы, точно соответствующие нескольким заданным параметрам одновременно.
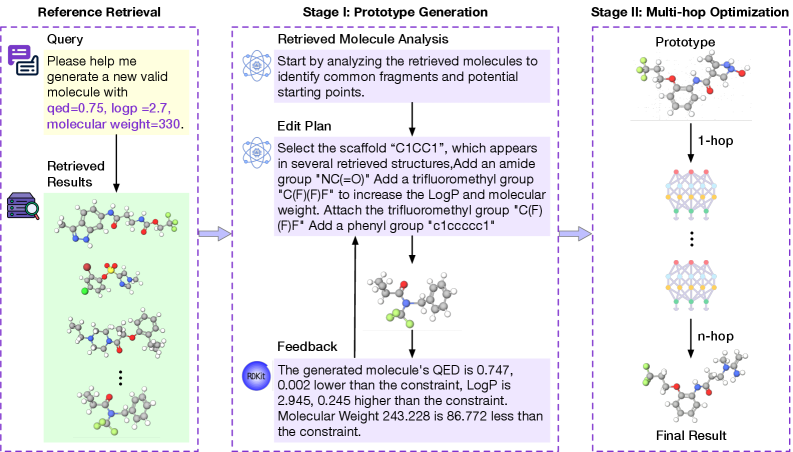
Представлена система M4olGen, использующая многоагентное обучение с подкреплением и большие языковые модели для фрагментного дизайна молекул с точным контролем над численными свойствами.
Создание молекул с заданными численными значениями нескольких физико-химических свойств представляет собой сложную задачу, требующую точного контроля и учета множества параметров. В данной работе представлена система M^4olGen: Multi-Agent, Multi-Stage Molecular Generation under Precise Multi-Property Constraints, — многоагентный, многоступенчатый фреймворк, использующий обучение с подкреплением и большие языковые модели для генерации молекул с точным контролем над несколькими численными свойствами одновременно. Предложенный подход позволяет достичь существенных улучшений в валидности и точности соответствия заданным свойствам, превосходя существующие графовые алгоритмы и крупные языковые модели. Каковы перспективы дальнейшей оптимизации и расширения возможностей M^4olGen для решения еще более сложных задач в области молекулярного дизайна и открытия новых материалов?
Химическое пространство: вызов многопараметрической оптимизации
Создание молекул с заданными свойствами является ключевой задачей в современной химии и фармацевтике, однако этот процесс часто осложняется противоречивыми требованиями. Например, повышение эффективности лекарственного препарата может негативно сказаться на его растворимости или селективности, что снижает его биодоступность и увеличивает риск побочных эффектов. Эта внутренняя борьба между желаемыми характеристиками представляет собой серьезную проблему для ученых, вынуждая их искать компромиссы или разрабатывать сложные стратегии, направленные на одновременное улучшение нескольких параметров. Успешное преодоление этих противоречий требует глубокого понимания взаимосвязей между молекулярной структурой и свойствами, а также применения передовых методов компьютерного моделирования и синтеза.
Традиционные методы молекулярного дизайна зачастую сталкиваются с серьезными трудностями при одновременной оптимизации нескольких ключевых характеристик. Например, повышение эффективности препарата (потентности) может негативно сказаться на его избирательности, то есть способности воздействовать только на целевую молекулу, избегая нежелательных побочных эффектов. Аналогично, улучшение растворимости, необходимое для биодоступности, нередко приводит к снижению стабильности молекулы. В результате, исследователи часто вынуждены идти на компромиссы, оптимизируя лишь одну или несколько характеристик в ущерб другим. Этот процесс требует значительных временных и ресурсных затрат, а также ограничивает возможности создания препаратов с оптимальным сочетанием свойств. Поиск эффективных решений, способных преодолеть эти ограничения, является одной из главных задач современной медицинской химии.
Эффективное исследование “химического пространства” требует принципиально новых подходов к согласованию конкурирующих требований при создании молекул. Традиционные методы, как правило, оптимизируют лишь отдельные характеристики, игнорируя взаимосвязи и компромиссы между ними. Инновационные стратегии, такие как многоцелевая оптимизация и применение алгоритмов машинного обучения, позволяют одновременно учитывать различные свойства, например, эффективность, селективность и растворимость. Эти методы способны находить молекулы, представляющие собой оптимальный баланс между желаемыми характеристиками, что особенно важно при разработке новых лекарственных препаратов и материалов с заданными свойствами. Вместо поиска единого «идеального» соединения, современные подходы направлены на выявление целого ряда перспективных кандидатов, обладающих приемлемыми значениями по всем ключевым параметрам.
Разработка молекул с заданными свойствами, особенно в контексте фармацевтики и материаловедения, требует одновременной оптимизации множества параметров — от эффективности и селективности до растворимости и стабильности. Традиционные подходы, как правило, фокусируются на оптимизации отдельных характеристик, игнорируя их взаимосвязь и потенциальные конфликты. В связи с этим, создание эффективной платформы, способной исследовать химическое пространство и находить компромиссы между различными требованиями, представляется задачей первостепенной важности. Такой фреймворк должен позволить исследователям не просто находить молекулы, обладающие желаемыми свойствами, но и прогнозировать их поведение в сложных системах, существенно ускоряя процесс разработки новых материалов и лекарственных средств. Реализация подобного подхода открывает возможности для создания инновационных продуктов с улучшенными характеристиками и расширенным спектром применения.
M4olGen: двухэтапный фреймворк для точного контроля молекул
Метод M4olGen представляет собой двухэтапный подход к генерации молекул, разработанный для преодоления ограничений традиционных методов. В отличие от одностадийных генеративных моделей, M4olGen разделяет процесс на этапы прототипирования и оптимизации. Это позволяет более эффективно исследовать химическое пространство и достигать более точного контроля над целевыми свойствами молекул. Традиционные методы часто сталкиваются с трудностями при одновременной оптимизации нескольких свойств, что приводит к компромиссам или неоптимальным результатам. Двухэтапная структура M4olGen позволяет сначала быстро создавать перспективные прототипы, а затем точно настраивать их свойства, повышая эффективность и точность процесса генерации.
На первом этапе, этапе прототипирования с использованием извлечения данных (Retrieval-Augmented Prototyping), M4olGen использует существующие базы данных молекулярных структур и их свойств для быстрой генерации перспективных прототипов. Вместо создания молекул с нуля, система идентифицирует существующие фрагменты и соединения, наиболее близкие к желаемым характеристикам, и комбинирует их, формируя начальные структуры. Этот подход значительно ускоряет процесс генерации, поскольку позволяет избежать ресурсоемких этапов de novo синтеза и предварительного отбора неперспективных вариантов. Использование существующих данных также способствует повышению реалистичности и химической выполнимости полученных прототипов.
Второй этап фреймворка M4olGen, оптимизация на уровне фрагментов на основе GRPO (Graph-based Retrosynthetic Prediction and Optimization), позволяет точно настраивать целевые свойства полученных молекулярных прототипов. GRPO использует граф-представление молекулы и применяет ретросинтетические предсказания для определения оптимальных модификаций фрагментов, направленных на улучшение таких характеристик, как QED, LogP, молекулярный вес (MW), а также значения HOMO и LUMO. Этот подход обеспечивает более гранулярный контроль над процессом генерации, позволяя достичь целевых свойств с высокой точностью и минимизировать отклонения от заданных параметров.
В ходе тестирования фреймворка M4olGen были получены следующие результаты по ключевым показателям молекулярных свойств: нормализованная общая ошибка составила 0.146 для QED, LogP и молекулярной массы (MW), и 0.155 для значений HOMO/LUMO. Данные показатели демонстрируют превосходный уровень контроля над процессом генерации молекул и более высокую точность предсказываемых свойств по сравнению с существующими методами. Полученные значения указывают на способность M4olGen генерировать молекулы с желаемыми характеристиками, минимизируя отклонения от целевых значений по указанным параметрам.
Фрагментный подход: оптимизация с использованием относительной политики оптимизации
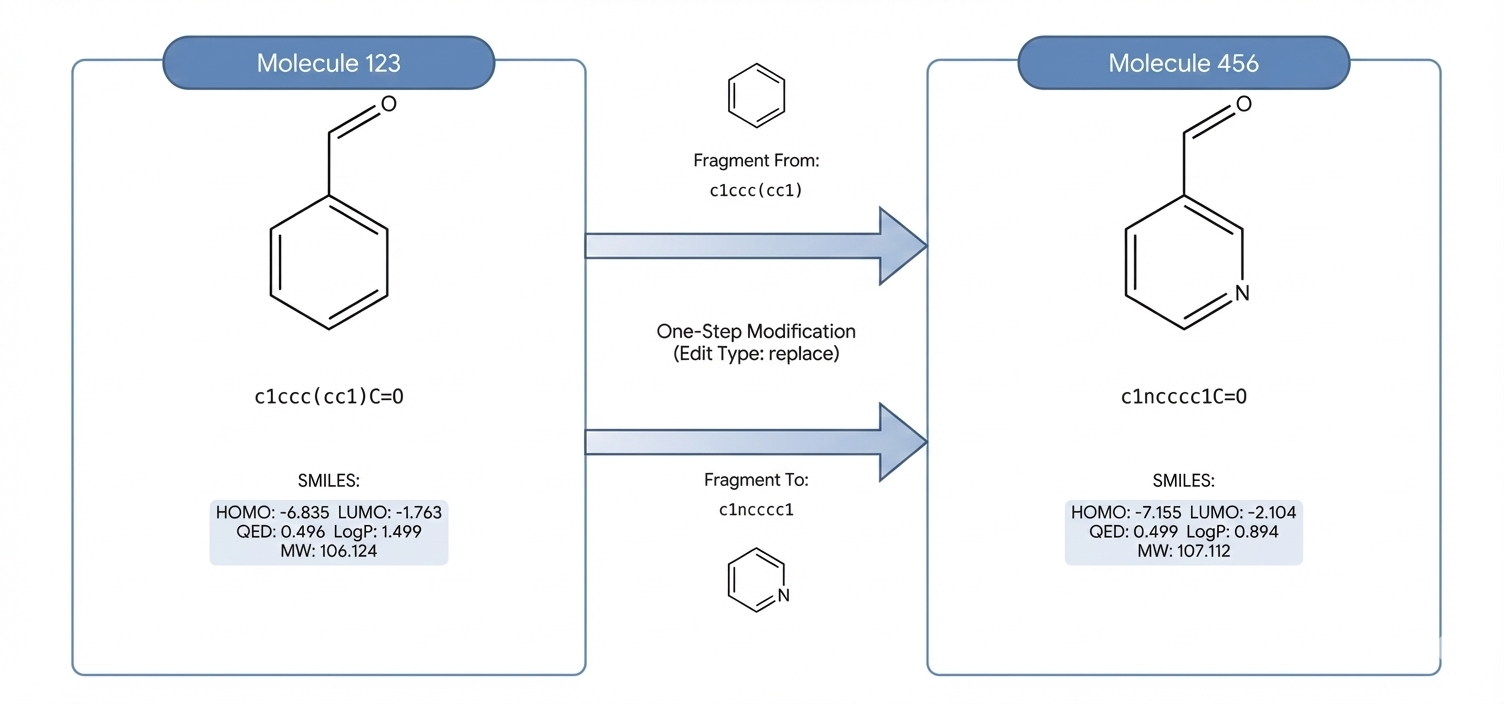
Оптимизация на основе Group Relative Policy Optimization (GRPO) и фрагментов предполагает интеллектуальное редактирование молекулярных структур посредством целенаправленных изменений, осуществляемых на уровне химически значимых строительных блоков — BRICS-фрагментов. Применяемый алгоритм GRPO позволяет производить изменения в структуре молекулы, опираясь на относительные предпочтения различных групп атомов, что обеспечивает более эффективный поиск оптимальных молекулярных свойств. Данный подход позволяет целенаправленно модифицировать отдельные фрагменты молекулы, минимизируя при этом нежелательные изменения в других частях структуры и сохраняя общую молекулярную целостность.
Метод оптимизации использует фрагменты BRICS — химически значимые строительные блоки, определяющие пространство допустимых изменений молекулярной структуры. Эти фрагменты представляют собой атомы или небольшие группы атомов, которые могут быть заменены или модифицированы без существенного нарушения общей химической структуры и свойств молекулы. Использование BRICS фрагментов позволяет алгоритму сосредоточиться на целевых изменениях, ограничивая пространство поиска и повышая эффективность оптимизации, в отличие от случайных изменений в пределах всей молекулы. Фактически, BRICS фрагменты служат предопределенными точками редактирования, обеспечивая более контролируемый и осмысленный процесс оптимизации молекулярных характеристик.
Оптимизация на уровне фрагментов позволяет системе вносить целенаправленные изменения в молекулярные структуры с минимальным влиянием на общие свойства молекулы. Вместо глобальных изменений всей структуры, алгоритм оперирует химически значимыми фрагментами (например, BRICS фрагментами) как строительными блоками. Такой подход позволяет модифицировать отдельные части молекулы, сохраняя при этом ее основные характеристики, такие как молекулярная масса и LogP. Это достигается за счет того, что изменения вносятся в пределах локализованных фрагментов, снижая вероятность нежелательных побочных эффектов и поддерживая структурную целостность молекулы.
Оптимизация с использованием Group Relative Policy Optimization (GRPO) позволила добиться значительного снижения ошибок в предсказываемых молекулярных свойствах. В ходе 3-хопового уточнения (3-hop refinement) наблюдалось уменьшение средней ошибки молекулярной массы (MW) на 84.9%, снизившись с приблизительно 63 до 10 единиц. Кроме того, ошибка в логарифме коэффициента распределения (LogP) была уменьшена на 70.7%. Данные результаты демонстрируют эффективность GRPO в оптимизации молекулярных структур с целью достижения заданных целевых значений свойств.
Валидация и уточнение: обеспечение свойств, близких к лекарственным
Сгенерированные молекулы подвергаются тщательной проверке на валидность с использованием RDKit — широко распространенного инструментария в области хемоинформатики. Этот комплексный набор функций позволяет оценить структурную корректность молекул, выявляя потенциальные ошибки или нереалистичные конфигурации. Помимо структурной проверки, RDKit рассчитывает ключевые свойства молекул, такие как молекулярный вес, число водородных связей и логарифм коэффициента распределения (logP). Эта проверка обеспечивает, что созданные структуры не только соответствуют химическим принципам, но и обладают характеристиками, необходимыми для дальнейшего исследования и потенциального применения в качестве лекарственных средств. Использование RDKit в процессе генерации молекул позволяет отсеять нежизнеспособные или неправдоподобные структуры на ранних стадиях, значительно повышая эффективность и результативность процесса разработки.
Для точного предсказания ключевых свойств молекул, таких как энергии высшей занятой молекулярной орбитали (ВЗМО) и низшей свободной молекулярной орбитали (НСМО), используется архитектура DimeNet++. Эта нейронная сеть, основанная на графовых свертках, позволяет эффективно учитывать пространственную структуру молекул и корреляции между атомами. Благодаря этому, предсказанные энергии ВЗМО и НСМО демонстрируют высокую точность, что критически важно для оценки реакционной способности, оптических свойств и других важных характеристик разрабатываемых соединений. Такой подход позволяет не только фильтровать нереалистичные или нежелательные молекулы на ранних этапах разработки, но и направленно проектировать соединения с заданными свойствами, что значительно ускоряет процесс поиска новых лекарственных средств и материалов.
Комбинация валидации и прогнозирования свойств играет ключевую роль в создании перспективных лекарственных кандидатов. Проверка с использованием инструментов, таких как RDKit, гарантирует, что сгенерированные молекулы соответствуют базовым химическим принципам и могут быть синтезированы в лабораторных условиях. Одновременно, точное предсказание характеристик, включая энергии высшей занятой и низшей свободной молекулярных орбиталей ( HOMO и LUMO ), позволяет отбирать соединения, обладающие желаемыми свойствами для взаимодействия с биологическими мишенями. Такой подход обеспечивает не только теоретическую обоснованность, но и практическую реализуемость разработанных молекул, значительно повышая вероятность успеха в процессе создания новых лекарственных препаратов.
Разработка M4olGen позволила достичь значительного превосходства над существующими подходами в области генерации молекул. В ходе сравнительного анализа было установлено, что M4olGen демонстрирует на 42,7% меньшую нормализованную общую ошибку по сравнению с наиболее мощной коммерческой моделью — GPT-4.1. Этот результат указывает на существенное улучшение точности предсказания свойств генерируемых соединений. Кроме того, M4olGen значительно превосходит базовую модель Graph GA, подтверждая эффективность предложенного подхода к решению задачи генерации новых молекул с заданными характеристиками. Такое достижение открывает новые возможности для ускорения процесса разработки лекарственных препаратов и создания материалов с улучшенными свойствами.
Исследование, представленное в данной работе, демонстрирует подход к генерации молекул, где сложная задача оптимизации множества свойств решается посредством многоагентной системы. Этот метод, использующий возможности больших языковых моделей и обучение с подкреплением, напоминает попытку взломать систему ограничений, наложенных на химическое пространство. Как однажды заметил Клод Шеннон: «Информация — это то, что уменьшает неопределенность». В контексте M4olGen, каждый агент, оптимизирующий определенное свойство, уменьшает неопределенность в процессе генерации молекул, стремясь к созданию соединений с заданными характеристиками. Таким образом, M4olGen представляет собой не просто генератор молекул, а инструмент для исследования границ возможного в химическом дизайне, подтверждая идею о том, что понимание системы позволяет её контролировать.
Куда Далее?
Представленная методология, несомненно, открывает новые горизонты в области молекулярного дизайна. Однако, следует признать: покорение многомерного пространства свойств — это не однократный взлом системы, а скорее непрерывный процесс реверс-инжиниринга. Оптимизация сразу нескольких параметров неизбежно приводит к компромиссам, и вопрос о том, как эффективно балансировать между ними, остаётся открытым. Необходимо углублённое исследование влияния различных архитектур агентов и стратегий обучения на стабильность и предсказуемость генерации.
Особый интерес представляет возможность интеграции M4olGen с другими вычислительными подходами, такими как квантово-химические расчёты или молекулярная динамика. Такой симбиоз позволит не только генерировать молекулы с заданными свойствами, но и оценивать их практическую реализуемость и стабильность в реальных условиях. Предстоит выяснить, насколько хорошо предложенный фрагментарный подход масштабируется для решения задач, требующих синтеза сложных, полициклических структур.
В конечном итоге, успех M4olGen, как и любого другого инструмента в руках исследователя, будет определяться не только его техническими возможностями, но и способностью задавать правильные вопросы. Поиск новых молекул — это, прежде всего, поиск новых закономерностей, и лишь умение выходить за рамки привычных представлений позволит раскрыть весь потенциал данной технологии.
Оригинал статьи: https://arxiv.org/pdf/2601.10131.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Квантовый Борьба: Китай и США на Передовой
- Квантовые симуляторы: проверка на прочность
- Квантовые нейросети на службе нефтегазовых месторождений
- Искусственный интеллект заимствует мудрость у природы: новые горизонты эффективности
- Интеллектуальная маршрутизация в коллаборации языковых моделей
- Квантовый скачок: от лаборатории к рынку
2026-01-17 02:53