Автор: Денис Аветисян
Исследователи разработали систему, способную предсказывать квантовые гамильтонианы молекул, используя только их SMILES-представления, без необходимости учитывать трехмерную структуру.
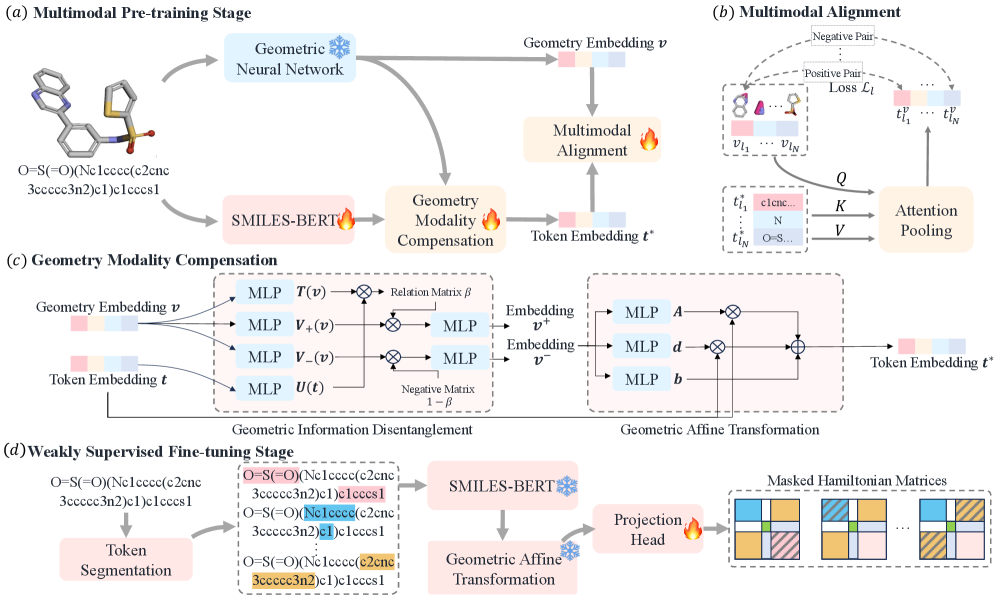
Предложенная модель MGAHam использует компенсацию модальностей и слабо контролируемое обучение для высокопроизводительного предсказания молекулярных гамильтонианов.
Вычисление квантового гамильтониана, определяющего электронную структуру молекул, является вычислительно затратной задачей, требующей обширных данных о геометрии и составе молекул. В данной работе, ‘Endowing Molecular Language with Geometry Perception via Modality Compensation for High-Throughput Quantum Hamiltonian Prediction’, предложен новый подход, позволяющий предсказывать гамильтониан непосредственно из упрощенного представления молекулы (SMILES) без использования трехмерных координат. Достигнуто это благодаря стратегии компенсации геометрической модальности, наделяющей языковое представление молекулы информацией о геометрии и обеспечивающей высокую точность предсказаний. Может ли разработанный метод значительно ускорить исследования в области химии и материаловедения, открывая новые возможности для высокопроизводительного моделирования молекул?
Молекулярное Предсказание: Вызов и Предел Расчётов
Точное предсказание молекулярных свойств, в частности матрицы Гамильтона, является фундаментальным для прогресса в области материаловедения и проектирования новых материалов. Матрица Гамильтона описывает энергетические уровни и взаимодействие электронов в молекуле, определяя её химические и физические характеристики. Способность точно рассчитывать эту матрицу позволяет исследователям предсказывать стабильность, реакционную способность и другие ключевые свойства материалов, значительно ускоряя процесс открытия новых соединений с заданными характеристиками. Например, предсказание электронной структуры позволяет оптимизировать материалы для солнечных батарей, сверхпроводников или катализаторов, минуя дорогостоящие и длительные экспериментальные исследования. HΨ = EΨ — это уравнение Шрёдингера, в котором H представляет собой матрицу Гамильтона, а Ψ — волновой функцией, описывающей состояние молекулы. Таким образом, точное моделирование матрицы Гамильтона открывает путь к целенаправленному дизайну материалов с уникальными и востребованными свойствами.
Традиционные методы, такие как теория функционала плотности (DFT), несмотря на свою точность в предсказании свойств молекул, сталкиваются с существенными вычислительными ограничениями. Вычисления, необходимые для моделирования даже относительно небольших систем, требуют значительных ресурсов и времени, что делает масштабные симуляции, необходимые для открытия и проектирования новых материалов, практически невозможными. С ростом сложности молекулярной структуры и размера рассматриваемой системы, вычислительная стоимость DFT растет экспоненциально, что ограничивает возможность исследования сложных химических процессов и материалов с высокой точностью. Это особенно актуально при моделировании систем, содержащих сотни или тысячи атомов, где вычислительные затраты становятся непомерно высокими, что стимулирует поиск альтернативных, более эффективных подходов к молекулярному моделированию.
Несмотря на многообещающие результаты, существующие геометрические нейронные сети сталкиваются с трудностями при эффективном моделировании сложной взаимосвязи между геометрией молекулы и её электронной структурой. Они часто испытывают ограничения в адекватном представлении квантово-механических эффектов, возникающих из-за сложного распределения электронов в молекуле. Это связано с тем, что традиционные архитектуры нейронных сетей не всегда способны эффективно кодировать информацию о трехмерной структуре молекулы и её влиянии на электронные свойства, такие как энергия и плотность электронов. В результате, точность предсказаний, основанных на этих сетях, может быть ограничена, особенно при работе со сложными молекулами и материалами, где геометрические детали играют решающую роль в определении их свойств.
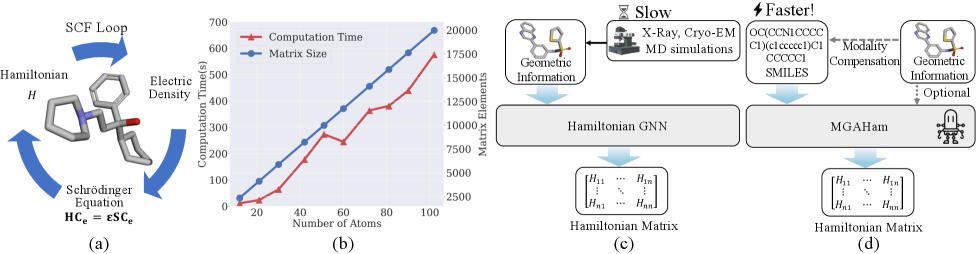
MGAHam: Новый Взгляд на Предсказание Гамильтониана
Модель MGAHam представляет собой новый многомодальный языковой подход, который позволяет предсказывать матрицу Гамильтона молекулы непосредственно из SMILES-строки, обходя необходимость в явном определении трехмерной геометрии. В отличие от традиционных методов, требующих предварительного расчета координат атомов, MGAHam оперирует непосредственно с текстовым представлением молекулы, что существенно упрощает процесс предсказания ее энергетических свойств. Это достигается за счет использования SMILES-строки как входных данных для построения языковой модели, способной извлекать информацию о структуре и связях внутри молекулы и преобразовывать ее в матрицу Гамильтона H, определяющую энергетические уровни системы.
Модель MGAHam использует комбинацию SMILES-BERT для получения токеновых вложений и QHNet для извлечения геометрической информации о молекуле. SMILES-BERT преобразует SMILES-строку, представляющую молекулу, в векторное представление, улавливающее ее химическую структуру. QHNet, в свою очередь, фокусируется на геометрических характеристиках молекулы. Для интеграции этих двух модальностей используется специализированный процесс Multimodal Alignment, который позволяет модели эффективно объединить информацию о структуре и геометрии, обеспечивая точное предсказание Гамильтониана молекулы. Этот подход позволяет избежать необходимости явного представления 3D-геометрии, упрощая и ускоряя процесс вычислений.
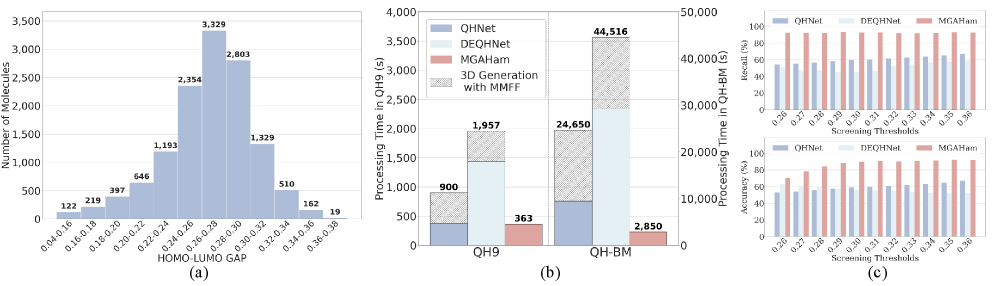
Архитектура MGAHam спроектирована с акцентом на вычислительную эффективность, что позволяет быстро предсказывать молекулярные свойства. На наборе данных QH9 модель демонстрирует ускорение в 115.42x по сравнению с расчетами, выполненными с использованием теории функционала плотности (DFT). Такая скорость достигается благодаря оптимизированной интеграции SMILES-BERT и QHNet, а также специализированному процессу выравнивания мультимодальных данных, что существенно снижает вычислительные затраты при сохранении точности предсказаний.
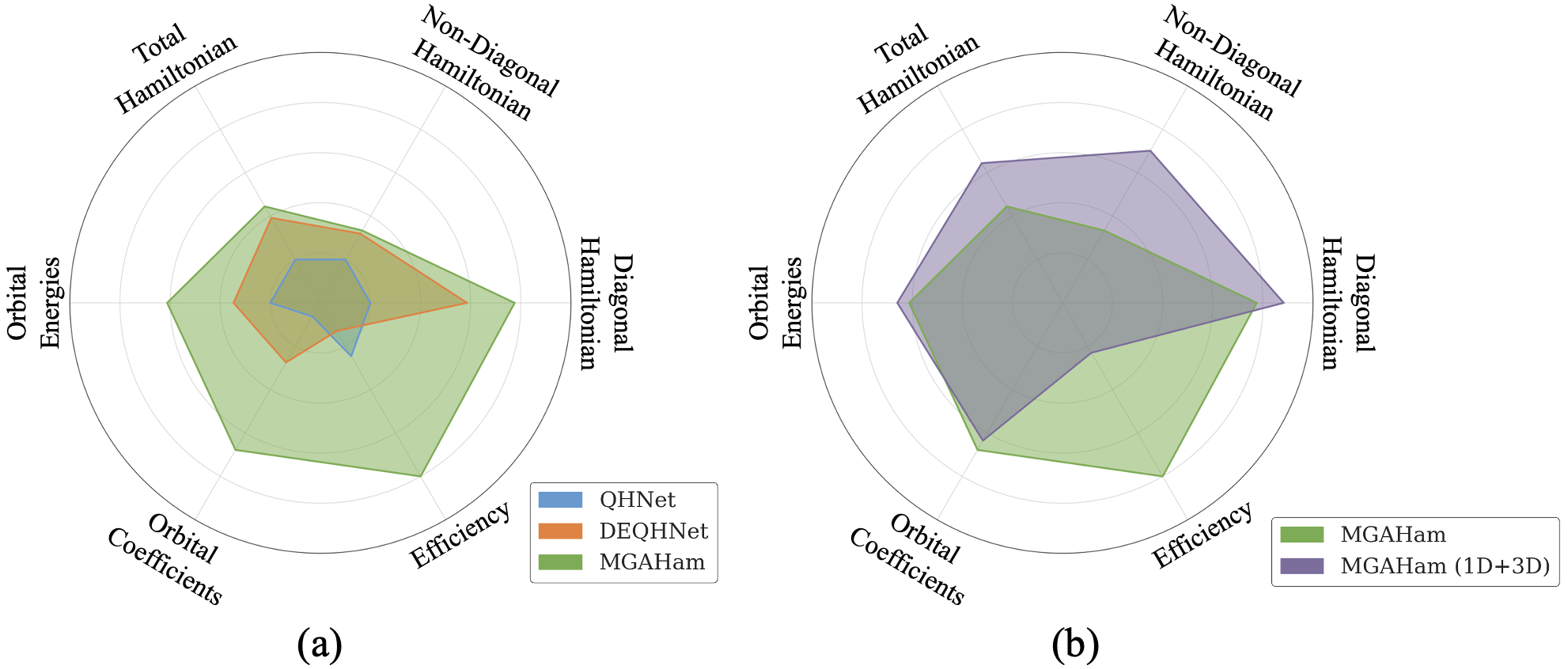
Валидация и Эффективность на Разнообразных Наборах Данных
Эффективность MGAHam была тщательно проверена на различных наборах данных, включающих MD17, QH9 и сложный бенчмарк QH-BM. Использование этих наборов позволило оценить обобщающую способность модели и её устойчивость к различным условиям. Набор MD17 представляет собой молекулярную динамику, QH9 — набор квантово-химических данных, а QH-BM — специально разработанный бенчмарк для оценки производительности моделей в сложных сценариях. Результаты валидации на этих данных демонстрируют надежность и применимость MGAHam для решения широкого круга задач в области молекулярного моделирования и квантовой химии.
Модель MGAHam демонстрирует улучшенную обобщающую способность благодаря применению слабо контролируемой тонкой настройки (Weakly Supervised Fine-Tuning). Этот метод позволяет эффективно обучать модель на замаскированных данных гамильтониана, что особенно важно для работы с неполной или зашумленной информацией. В процессе обучения модель использует доступные данные для прогнозирования скрытых частей гамильтониана, что способствует более надежному и точному моделированию молекулярных систем в различных условиях и на разнообразных наборах данных.
При валидации на наборе данных MD17 для молекулы воды, модель MGAHam достигла ошибки Гамильтона (H, Eh) в размере 1.73 \times 10^{-2}, продемонстрировав превосходство над всеми базовыми моделями. Дополнительно, при использовании модификации MGAHam (1D+3D) на бенчмарке QH-BM, наблюдалось снижение ошибки Гамильтона на 38.3% по сравнению со стандартной версией MGAHam.
Влияние на Открытие Материалов и Проектирование Электролитов
Точное предсказание ширины запрещенной зоны — разницы энергий высшей заполненной молекулярной орбитали (ВЗМО) и низшей свободной молекулярной орбитали (НСМО) — при помощи метода MGAHam имеет решающее значение для понимания и оптимизации свойств электролитов, используемых в литий-ионных аккумуляторах. Ширина запрещенной зоны напрямую влияет на электрохимическую стабильность и ионную проводимость электролита, определяя его способность эффективно транспортировать ионы лития, не подвергаясь при этом нежелательным реакциям разложения. MGAHam позволяет точно рассчитывать эти значения, предоставляя возможность предсказывать поведение электролитов в различных условиях и, следовательно, проектировать составы с улучшенной производительностью и долговечностью. Это особенно важно для разработки электролитов нового поколения, способных работать при экстремальных температурах или с повышенным напряжением, что является ключевым фактором для увеличения энергоемкости и срока службы аккумуляторов.
Возможность точного прогнозирования энергетических уровней молекул, в частности, щели между высшей занятой и низшей незанятой молекулярными орбиталями, открывает новые горизонты в разработке электролитов для литий-ионных аккумуляторов. Это позволяет целенаправленно конструировать химический состав электролитов, оптимизируя их характеристики для повышения эффективности и стабильности работы аккумулятора. Благодаря такому подходу, исследователи получают возможность предсказывать, как различные комбинации растворителей и солей повлияют на ключевые параметры электролита, например, на ионную проводимость и электрохимическую стабильность. В результате, процесс создания новых электролитов становится более рациональным и менее затратным, поскольку значительно сокращается число необходимых экспериментальных испытаний, а также появляется возможность проектировать материалы с заданными свойствами, отвечающие конкретным требованиям будущих аккумуляторов.
Разработка MGAHam значительно ускоряет процесс открытия новых материалов, открывая перспективы для инноваций не только в области хранения энергии, но и в катализе, а также других ключевых областях науки и техники. Точное предсказание энергии LUMO позволяет проводить эффективный скрининг составов электролитов, учитывая свойства растворителей и солей, что позволяет целенаправленно создавать новые формулы с улучшенными характеристиками. Такой подход позволяет сократить время и затраты на экспериментальные исследования, фокусируясь на наиболее перспективных кандидатах и тем самым стимулируя появление материалов нового поколения с повышенной эффективностью и стабильностью.
Представленная работа демонстрирует стремление обойти необходимость в трёхмерных координатах молекул при предсказании гамильтонианов, используя лишь SMILES-строки. Это, конечно, элегантно, но напоминает о вечной гонке за упрощением, где каждая «революционная» технология завтра станет техдолгом. Попытки компенсировать недостающую модальность посредством обучения кажутся разумными, однако всегда есть риск, что при увеличении нагрузки на систему, теоретическая точность уступит место практическим ограничениям. Как говорил Алан Тьюринг: «Можно только надеяться, что компьютеры в конечном итоге научатся делать всё». Но пока они этого не сделали, стоит помнить, что иногда лучше монолит, чем сто микросервисов, каждый из которых врёт.
Что дальше?
Представленный подход, безусловно, изящен. Освободить предсказание молекулярных гамильтонианов от необходимости в трёхмерных координатах — это, конечно, удобно. Но не стоит забывать, что реальный мир, в отличие от красивых тензоров, любит подбрасывать сюрпризы. Каждый новый, чуть более сложный класс молекул неизбежно выявит слабые места в представлении SMILES-строк, заставив систему искать обходные пути. И эти пути, вероятнее всего, окажутся не такими элегантными, как хотелось бы.
Неизбежно возникнет вопрос о масштабируемости. Ускорение вычислений — это хорошо, но что произойдёт, когда количество молекул, требующих анализа, достигнет астрономических величин? Появится потребность в ещё более компактных представлениях, ещё более хитрых алгоритмах. И тогда возникнет соблазн вернуться к старым добрым трюкам с оптимизацией, только уже на новых, более изощрённых платформах. Тесты, как всегда, останутся лишь формой надежды, а не гарантией.
В конечном итоге, вся эта гонка за ускорением — лишь отсрочка неизбежного. Продакшен всегда найдёт способ сломать даже самую изящную теорию. И тогда придётся снова копаться в низкоуровневых деталях, оптимизировать циклы и бороться с погрешностями. Каждая «революционная» технология завтра станет техдолгом, а квантовая химия, как и любая другая область, продолжит жить по своим, непредсказуемым законам.
Оригинал статьи: https://arxiv.org/pdf/2601.15786.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Квантовые Заметки: Прогресс и Парадоксы
- Звуковая фабрика: искусственный интеллект, создающий музыку и речь
- Квантовые нейросети на службе нефтегазовых месторождений
- Квантовые симуляторы: точное вычисление энергии основного состояния
- Кватернионы в машинном обучении: новый взгляд на обработку данных
- Кванты в Финансах: Не Шутка!
- Квантовые сети для моделирования молекул: новый подход
- Ускорение оптимального управления: параллельные вычисления в QPALM-OCP
- Миллиардные обещания, квантовые миражи и фотонные пончики: кто реально рулит новым золотым веком физики?
- Функциональные поля и модули Дринфельда: новый взгляд на арифметику
2026-01-24 21:52