Автор: Денис Аветисян
Исследование демонстрирует, что комбинация нейронных квантовых состояний и метода выбранного взаимодействия конфигураций превосходит традиционные методы вариационного Монте-Карло в задачах квантовой химии.
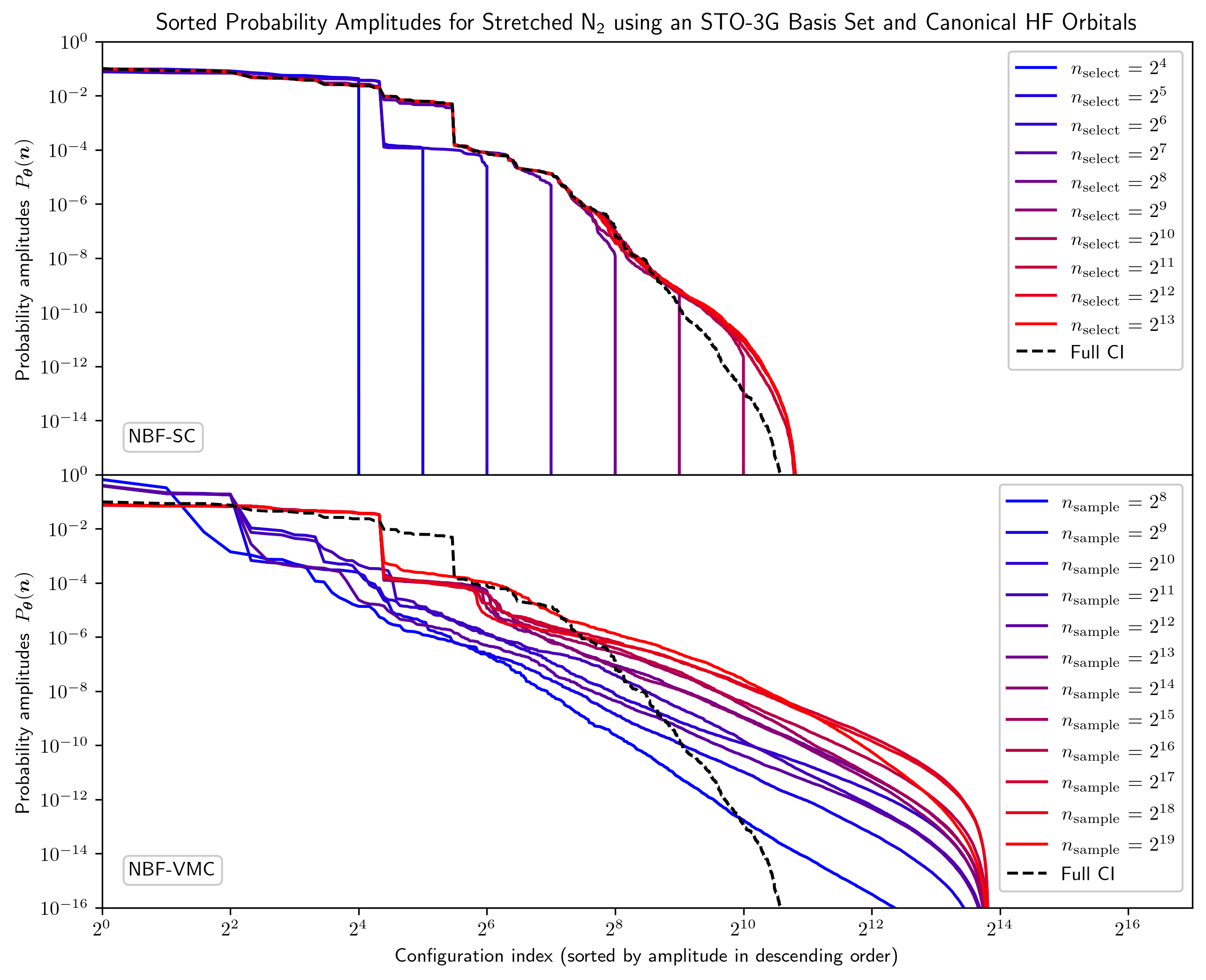
Предложенный подход NQS-SC обеспечивает повышенную точность и масштабируемость при вычислении электронных состояний, превосходя NQS-VMC.
Несмотря на значительные успехи в разработке нейронных сетей для квантовой химии, точное и эффективное вычисление электронных энергий остается сложной задачей. В настоящей работе, посвященной исследованию ‘Neural Quantum States Based on Selected Configurations’, предложен и всесторонне изучен новый подход, сочетающий нейронные квантовые состояния с методом выбранной конфигурации (NQS-SC). Показано, что NQS-SC значительно превосходит традиционный метод вариационного Монте-Карло (NQS-VMC) в точности энергии и коэффициентов волновой функции, особенно для молекул со статической корреляцией. Какие перспективы открываются для дальнейшего развития гибридных методов, объединяющих преимущества NQS с более продвинутыми теориями возмущений для описания динамической корреляции?
Преодолевая Экспоненциальную Сложность: К Необходимости Нейронных Квантовых Состояний
Решение многочастичного уравнения Шрёдингера представляет собой фундаментальную проблему в квантовой химии и физике конденсированного состояния, однако его вычислительная сложность экспоненциально возрастает с увеличением числа частиц в системе. Это означает, что точное моделирование даже относительно небольших молекул или материалов становится практически невозможным, ограничивая возможности детального изучения их свойств и предсказания химических реакций. Традиционные методы, такие как метод Хартри-Фока и полная конфигурационная интеграция, демонстрируют ограниченную применимость из-за этой вычислительной неразрешимости, что создает значительные препятствия для прогресса в области материаловедения и разработки новых лекарственных препаратов. Несмотря на значительные успехи в области вычислительной мощности, эта проблема остается актуальной и требует поиска новых, инновационных подходов к решению квантовомеханических задач.
Традиционные методы решения квантово-механических задач, такие как метод Хартри-Фока и метод полной конфигурации, сталкиваются с серьезными ограничениями при моделировании сложных молекулярных систем. Суть проблемы заключается в экспоненциальном росте вычислительных затрат с увеличением числа частиц, описываемых в модели. Это означает, что для каждой добавленной частицы, необходимая мощность вычислений увеличивается не линейно, а в разы, что делает точное моделирование даже умеренно сложных молекул практически невозможным на современных компьютерах. В результате, прогресс в области химического открытия и разработки новых материалов существенно замедляется, поскольку исследователи ограничены в своей способности предсказывать свойства и поведение сложных химических соединений с высокой точностью. O(N!) — именно такая сложность возникает при использовании метода полной конфигурации для N-электронной системы, что делает его неприменимым для сколько-нибудь значимых молекул.
Нейронные квантовые состояния (НКС) представляют собой перспективный подход к моделированию сложных квантовых систем, используя возможности нейронных сетей для представления волновых функций. В отличие от традиционных методов, таких как метод Хартри-Фока и полная конфигурационная интеграция, которые страдают от экспоненциального роста вычислительных затрат с увеличением размера системы, НКС позволяют аппроксимировать волновые функции с использованием параметризованных нейронных сетей. Это позволяет эффективно описывать корреляции между частицами, которые критически важны для точного моделирования молекул и материалов. Благодаря способности нейронных сетей к обобщению и интерполяции, НКС могут потенциально преодолеть ограничения традиционных методов и открыть новые возможности для квантово-химических расчетов и открытия новых материалов с заданными свойствами. Такой подход позволяет исследовать системы, недоступные для стандартных вычислительных методов, и получить более глубокое понимание квантовых явлений.

Вариационный Монте-Карло и Стратегии Сэмплирования: Фундамент Точных Расчетов
Метод вариационного Монте-Карло (ВМК) представляет собой вычислительную схему оценки энергии волновой функции N-частичной квантовой системы путём статистической выборки конфигураций из этой волновой функции. В рамках ВМК, энергия вычисляется как среднее значение оператора Гамильтона по ансамблю случайных конфигураций, взвешенных вероятностью, определяемой квадратом модуля волновой функции |ψ(R)|², где R представляет собой вектор координат всех частиц системы. Точность оценки энергии напрямую зависит от эффективности выборки конфигураций, отражающих распределение вероятностей, заданное волновой функцией, и от размера ансамбла выбранных конфигураций.
Первые методы вариационного Монте-Карло (ВМК), такие как сэмплинг Metropolis-Hastings и авторегрессионный сэмплинг, представляли собой начальные подходы к исследованию конфигурационного пространства. Алгоритм Metropolis-Hastings, основанный на построении Марковской цепи, позволяет генерировать последовательность конфигураций, принимая или отклоняя предложенные изменения с вероятностью, зависящей от энергии и функции предложения. Авторегрессионный сэмплинг, в свою очередь, строит конфигурацию последовательно, определяя каждую координату на основе условного распределения, заданного предыдущими координатами. Оба метода обеспечивали возможность оценки энергии волновой функции, однако их эффективность была ограничена, особенно при работе с системами высокой размерности и сложными корреляциями между частицами.
Эффективность выборки является критически важным фактором для точной оценки энергии в методе вариационного Монте-Карло, особенно в системах с высокой размерностью. С увеличением числа степеней свободы системы, объем пространства конфигураций экспоненциально возрастает, что затрудняет эффективное исследование пространства и требует значительного увеличения числа выборок для достижения заданной точности. Неэффективные методы выборки приводят к высокой корреляции между последовательными выборками, что снижает статистическую значимость результатов и требует еще большего количества вычислений для получения надежной оценки энергии E. Поэтому, разработка и применение продвинутых стратегий выборки, способных эффективно исследовать пространство конфигураций и уменьшить корреляцию между выборками, является ключевой задачей для повышения точности и снижения вычислительных затрат при использовании метода вариационного Монте-Карло.
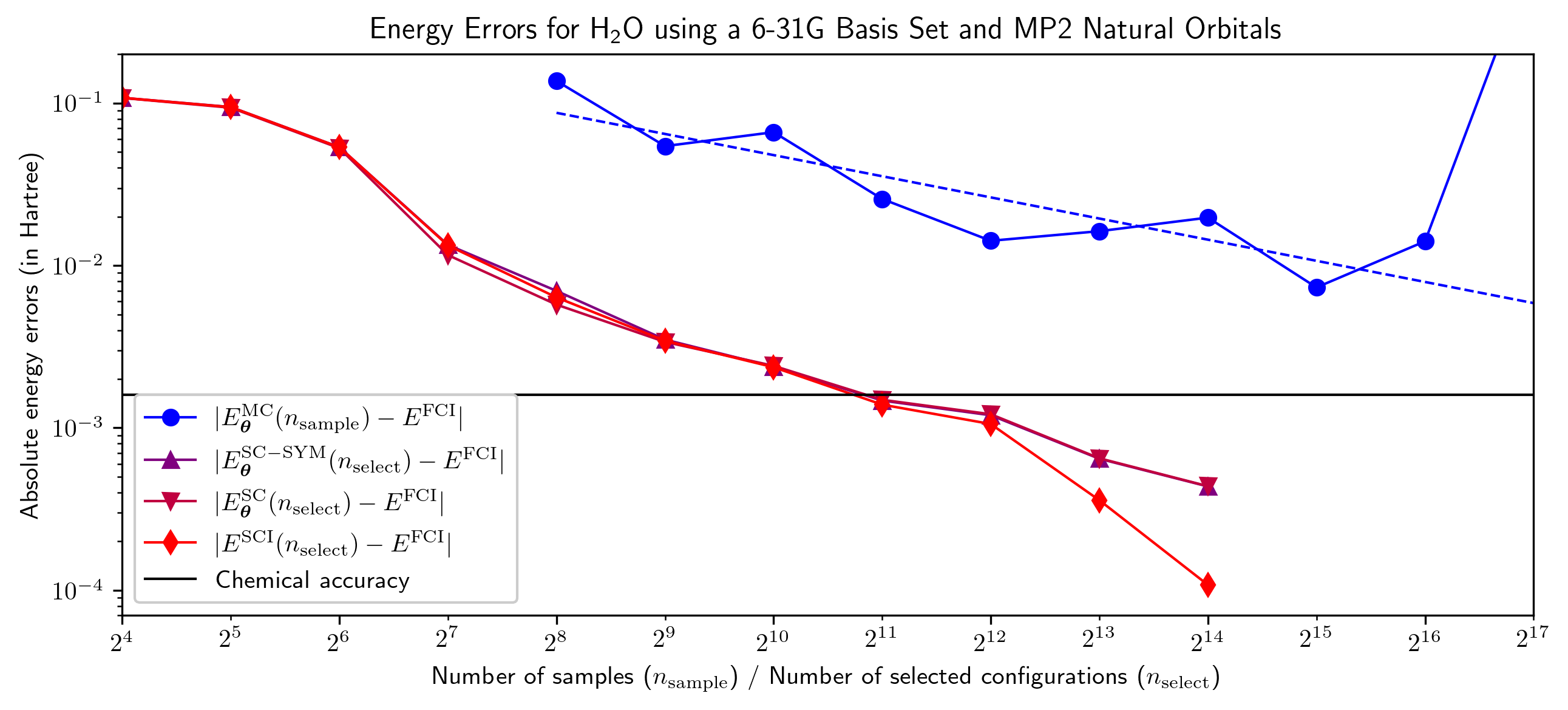
NQS-SC: Отобранные Конфигурации — Ключ к Эффективности Расчетов
Метод NQS-SC является расширением методов Selected Configuration Interaction (SCI) и использует NQS (Natural Quantum States) в качестве базового варианта волновой функции. В отличие от традиционных SCI, где выбор конфигураций основывается на энергии, NQS-SC использует принципы естественных квантовых состояний для определения наиболее значимых конфигураций, что позволяет достичь высокой точности при значительно меньшем количестве учитываемых конфигураций. По сути, NQS-SC заменяет стандартный выбор конфигураций в SCI на выбор, оптимизированный с использованием |\Phi_i\rangle — естественных квантовых состояний, полученных из волновой функции, построенной по принципу минимальной энергии.
Метод NQS-SC осуществляет стратегический отбор подмножества конфигураций, основываясь на информации, выходящей за рамки исходного волнового анзаца NQS. Это позволяет достичь химической точности при использовании менее 1% от общего конфигурационного пространства. Отбор конфигураций производится таким образом, чтобы обеспечить адекватное описание корреляционных эффектов, избегая при этом экспоненциального роста вычислительных затрат, характерного для полных методов конфигурационного взаимодействия. Эффективность подхода заключается в использовании дополнительных критериев отбора, позволяющих идентифицировать наиболее важные конфигурации, вносящие значительный вклад в полную энергию системы.
В методе NQS-SC начальный выбор конфигураций осуществляется на основе волновой функции Хартри-Фока, что позволяет эффективно сократить пространство конфигураций для дальнейших вычислений. Использование решения Хартри-Фока в качестве «затравки» обеспечивает разумный начальный набор конфигураций, необходимых для построения более точного приближения. Для оценки энергии полученных конфигураций и проведения итеративных расчетов активно используется программный пакет PyCI, предоставляющий инструменты для эффективной работы с конфигурационными взаимодействиями и оптимизации волновой функции.
![Решения NBF, представленные на рисунке 1, демонстрируют, что ошибка энергии <span class="katex-eq" data-katex-display="false">|E^{\mathrm{SCI}}(n\_{\mathrm{select}})-E^{\mathrm{FCI}}|[ /latex] снижается до приблизительно [latex]5\cdot 10^{-{11}}[ /latex] Хартри и остается на этом уровне при дальнейшем увеличении [latex]n\_{\mathrm{select}}.</span>](https://arxiv.org/html/2602.12993v1/figure2.png)
Универсальность NQS-SC: Подтверждение Эффективности на Различных Молекулах
Метод NQS-SC продемонстрировал свою универсальность, успешно применяясь к широкому спектру молекул, включая как простые диатомные системы, такие как N_2 и H_2O, так и ионные соединения, например, LiCl, а также более сложные органические молекулы, такие как C_2H_4, и неорганическое соединение Li_2O. Этот успех в применении к разнообразным химическим системам подтверждает способность NQS-SC эффективно описывать различные типы химических связей и электронных структур, что делает его ценным инструментом для квантово-химических расчетов и моделирования свойств веществ.
Проведенные вычисления с использованием различных базисных наборов, от минимального STO-3G до обширного 6-311G, демонстрируют замечательную адаптивность разработанного метода NQS-SC к различным уровням теоретического подхода. Такая гибкость позволяет исследователям эффективно применять данный метод для изучения молекул с разной сложностью и точностью, в зависимости от доступных вычислительных ресурсов и требуемой степени детализации. Способность метода сохранять точность и эффективность при использовании как простых, так и сложных базисных наборов подчеркивает его универсальность и делает его ценным инструментом в арсенале квантовой химии и материаловедения.
Исследования молекулы азота в растянутом состоянии продемонстрировали высокую точность метода NQS-SC, достигшую минимальной погрешности в 11.4 \mu Ha при использовании nselect = 2^{10}. Особенно примечательно, что для достижения так называемой "химической точности" методу NQS-SC требуется менее 1% от всего конфигурационного пространства, что значительно превосходит показатели метода NQS-VMC. Данное свойство указывает на эффективность и вычислительную экономичность NQS-SC при моделировании сложных молекулярных систем, открывая перспективы для его применения в широком спектре химических задач, где точные расчеты энергии являются критически важными.
В основе разработанного подхода лежит использование пакета PySCF для получения высокоточных эталонных решений посредством метода полной конфигурационной интеракции (Full Configuration Interaction, FCI). Это обеспечивает надежность и достоверность полученных результатов, поскольку FCI, хотя и вычислительно затратный, признан «золотым стандартом» для расчетов электронных структур. Использование PySCF позволяет эффективно реализовать FCI-расчеты и служить отправной точкой для оценки точности и эффективности нового метода - NQS-SC. Такой подход гарантирует, что любые улучшения, достигнутые с помощью NQS-SC, действительно отражают повышение точности расчетов, а не связаны с погрешностями в эталонных данных. Надежность эталонных расчетов, полученных с помощью PySCF, является ключевым фактором для валидации и широкого применения NQS-SC к различным молекулам и системам.
Будущее Нейронного Квантового Моделирования: Открывая Новые Горизонты
Современные вычислительные платформы, такие как NetKet, JAX и Flax, значительно упрощают процесс обучения нейронных сетей для квантового моделирования (NQS). Эти фреймворки предоставляют автоматизированные инструменты для оптимизации параметров нейронных сетей, что позволяет достигать более высокой точности и эффективности в симуляциях сложных квантовых систем. В отличие от традиционных методов, требующих ручной настройки и значительных вычислительных ресурсов, данные платформы позволяют исследователям сосредоточиться на физической задаче, а не на технических деталях реализации алгоритма. Автоматизация оптимизации не только ускоряет процесс разработки, но и открывает возможности для исследования более сложных и масштабных квантовых систем, ранее недоступных для моделирования.
Архитектуры Neural Backflow представляют собой перспективный подход к повышению выразительности квантового моделирования с помощью нейронных сетей. В традиционных методах, базисные функции, описывающие поведение электронов в молекулах, задаются фиксированно. Neural Backflow позволяет нейронной сети динамически адаптировать коэффициенты этих базисных функций, фактически “обучая” оптимальное представление волновой функции системы. Это достигается путем введения дополнительного преобразования координат, определяемого нейронной сетью, которое позволяет более эффективно описывать корреляции между электронами. В результате, повышается точность расчетов даже для сложных молекулярных систем, что открывает новые возможности для моделирования материалов и разработки лекарственных препаратов, где точное описание электронных взаимодействий играет ключевую роль. Такой подход позволяет преодолеть ограничения традиционных методов и приблизиться к решению задач, ранее считавшихся недоступными для квантового моделирования.
Сочетание передовых разработок в области нейросетевых квантовых симуляций открывает перспективы для точного и эффективного моделирования сложных химических систем. В частности, интеграция фреймворков, таких как NetKet, JAX и Flax, с архитектурами Neural Backflow позволяет не только автоматизировать процесс оптимизации, но и значительно повысить выразительность симуляций за счет обучения адаптивным коэффициентам орбиталей. Такой подход позволяет решать задачи, которые ранее были недоступны из-за вычислительных ограничений, что, в свою очередь, стимулирует инновации в материаловедении и фармацевтике. В перспективе это может привести к созданию новых материалов с заданными свойствами и разработке принципиально новых лекарственных препаратов, моделирование которых становится возможным с беспрецедентной точностью и скоростью.
Исследование, представленное в данной работе, демонстрирует значительный прогресс в области вычислительной квантовой химии. Подход, объединяющий Neural Quantum States и Selected Configuration Interaction (NQS-SC), превосходит традиционный Variational Monte Carlo (NQS-VMC) по точности и масштабируемости. Этот результат подчеркивает важность поиска элегантных и доказуемых алгоритмов для решения сложных задач. Как однажды заметил Григорий Перельман: «Математика - это язык Бога». В данном случае, точность вычислений, достигаемая благодаря NQS-SC, является прямым следствием математической чистоты и строгости используемых методов, что позволяет надежно рассчитывать электронные структуры и приближаться к истинному решению.
Что Дальше?
Представленные результаты, демонстрирующие превосходство подхода Neural Quantum States в сочетании с Selected Configuration Interaction над традиционным Variational Monte Carlo, не являются, конечно, окончательным ответом. Скорее, это очередное подтверждение давно известного факта: замена явных математических конструкций на неявно заданные аппроксимации требует чрезвычайной осторожности. Доказательство сходимости и корректности такого рода алгоритмов - задача, не имеющая тривиального решения, и требующая строгого математического анализа, а не просто демонстрации успешной работы на ограниченном наборе тестовых примеров.
Очевидным направлением дальнейших исследований представляется разработка методов, позволяющих оценивать априорную погрешность, вносимую нейронной сетью в решение уравнения Шрёдингера. Недостаточно просто добиться высокой точности; необходимо понимать, насколько надежны полученные результаты и в каких случаях они могут быть неверными. В частности, вопрос об устойчивости к изменениям архитектуры нейронной сети и параметров обучения остается открытым.
Следует признать, что текущий подход, как и большинство методов вычислительной квантовой химии, сталкивается с экспоненциальным ростом вычислительных затрат при увеличении размера исследуемой системы. Таким образом, поиск более эффективных алгоритмов, позволяющих преодолеть это ограничение, остается фундаментальной задачей. И, как всегда, истинная элегантность решения будет заключаться не в его сложности, а в математической чистоте и доказуемости.
Оригинал статьи: https://arxiv.org/pdf/2602.12993.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Внимание на границе: почему трансформеры нуждаются в «поглотителях»
- Внимание в сети: Новый подход к ускорению больших языковых моделей
- Искусственный нос будущего: как квантовая механика и машинное обучение распознают запахи
- Химический синтез под контролем искусственного интеллекта: новые горизонты
- Когда большая языковая модель молчит: как избежать галлюцинаций при ответе на вопросы?
- Свет под контролем: новые горизонты чиповой фотоники
- Квантовая телепортация в новых измерениях: топологические изоляторы
- Звук в коде: новая эра токенизации аудио
- Малые модели — большие возможности: Искусственный интеллект на производстве
- Видеовопросы и память: Искусственный интеллект на грани
2026-02-16 08:46