Автор: Денис Аветисян
Исследователи предлагают метод многоцелевой оптимизации и квантово-классической гибридизации для повышения точности и эффективности моделей, предсказывающих энергии и силы в органических и неорганических соединениях.
Многоцелевая оптимизация гиперпараметров и квантово-классические гибридные модели для обучения межмолекулярных потенциалов Allegro.
Повышение точности и скорости расчётов в молекулярной динамике часто представляет собой компромисс между этими параметрами. В данной работе, посвященной ‘Multi-objective optimization and quantum hybridization of equivariant deep learning interatomic potentials on organic and inorganic compounds’, предложен подход к многокритериальной оптимизации гиперпараметров модели Allegro, использующей E(3)-эквивариантные нейронные сети для предсказания межатомных потенциалов. Разработаны варианты архитектуры, включающие как классические многослойные персептроны, так и гибридные квантово-классические слои, что позволило добиться улучшения показателей точности и эффективности. Каковы перспективы дальнейшего развития гибридных моделей для моделирования сложных химических систем и расширения применимости методов машинного обучения в материаловедении?
Пределы Существующих MLIP: Компромисс Между Точностью и Вычислительными Затратами
Традиционные потенциалы межатомного взаимодействия, разработанные с использованием методов машинного обучения, часто сталкиваются с проблемой компромисса между точностью и вычислительными затратами. Стремление к высокой точности предсказаний требует более сложных моделей и, как следствие, значительного увеличения времени вычислений. Это особенно критично при моделировании больших систем или проведении длительных молекулярно-динамических симуляций, что существенно замедляет процесс открытия новых материалов. Несмотря на значительные успехи в области машинного обучения, поиск оптимального баланса между этими двумя факторами остается сложной задачей, ограничивающей возможности применения MLIP в масштабных исследованиях и затрудняющей эффективное прогнозирование свойств материалов с требуемой достоверностью.
Существующие модели машинного обучения межатомных потенциалов часто демонстрируют ограниченную способность точно описывать сложные химические окружения, что приводит к ненадежным прогнозам свойств материалов. Проблема заключается в том, что традиционные подходы, как правило, опираются на упрощенные представления об электронной структуре и взаимодействиях между атомами, не учитывая в полной мере эффекты многоатомных связей, поляризации и других квантово-механических явлений. Это особенно заметно при моделировании систем с высокой степенью беспорядка, дефектами или сложными химическими составами, где даже небольшие отклонения в описании химического окружения могут существенно повлиять на предсказываемые свойства, такие как энергия, сила и динамика. В результате, использование таких моделей может приводить к ошибочным выводам при поиске и разработке новых материалов с заданными характеристиками, подчеркивая необходимость разработки более совершенных подходов, способных адекватно учитывать сложность реальных химических сред.
Обеспечение эквивариантности — сохранения непротиворечивости при вращениях и трансляциях — является фундаментальным требованием для получения физически достоверных предсказаний в моделировании материалов. Неспособность модели учитывать симметрии системы приводит к нефизичным результатам и требует значительных вычислительных ресурсов для компенсации этих ошибок. Достижение истинной эквивариантности в моделях межатомного потенциала представляет собой сложную задачу, поскольку требует разработки алгоритмов, способных эффективно учитывать все возможные преобразования системы без существенного увеличения вычислительной нагрузки. Современные подходы часто используют сложные архитектуры нейронных сетей или специализированные функции, чтобы приблизиться к желаемой эквивариантности, однако компромисс между точностью, скоростью и способностью обобщать на новые системы остается актуальной проблемой в области вычислительной материаловедения.
Allegro: Основа, Адаптированная к Симметрии
Модель Allegro является развитием фреймворка NequiP и демонстрирует значительное повышение производительности за счет явного включения вращательной и поступательной симметрий. В отличие от традиционных подходов, не учитывающих инвариантность относительно поворотов и сдвигов, Allegro использует принципы симметрии для создания более эффективных представлений молекулярных структур. Это достигается путем преобразования входных данных таким образом, чтобы предсказания оставались согласованными независимо от ориентации и положения атомов в пространстве, что позволяет модели обобщать знания на новые, ранее не встречавшиеся химические среды с повышенной точностью и скоростью обучения. E(3) группа используется для обеспечения такой инвариантности.
Модель Allegro использует группу E(3) для обеспечения инвариантности предсказаний относительно ориентации и положения атомов в пространстве. Группа E(3) представляет собой группу Ли, описывающую все возможные комбинации вращений и трансляций в трехмерном пространстве. Применение принципов E(3)-симметрии позволяет Allegro обрабатывать входные данные, не зависящие от системы координат, что приводит к более устойчивым и обобщаемым предсказаниям свойств молекул и материалов. Фактически, это означает, что модель выдаст одинаковый результат для одной и той же молекулы, независимо от того, как она повернута или перемещена в пространстве, что существенно повышает надежность и эффективность расчетов.
Адаптация к симметриям в модели Allegro обеспечивает более эффективное обучение и улучшенную обобщающую способность к новым, ранее не встречавшимся химическим средам. Использование информации о симметриях молекулярной структуры позволяет модели извлекать больше полезной информации из ограниченного количества данных, снижая потребность в больших обучающих выборках. Это достигается за счет того, что модель учится распознавать и использовать инвариантные характеристики молекул, которые не меняются при поворотах и трансляциях, что позволяет ей делать более точные прогнозы для различных конфигураций атомов и молекул, даже тех, которые отличаются от тех, на которых она была обучена. По сути, симметрия выступает в качестве регуляризатора, упрощающего задачу обучения и повышающего устойчивость модели к изменениям в структуре входных данных.
Расширение Экспрессивности: Варианты Allegro++
Для увеличения емкости модели Allegro была разработана модификация Allegro++MLP, в которой добавлены дополнительные многослойные перцептроны (MLP). Включение MLP-слоев позволяет модели изучать более сложные зависимости в данных и повышает ее способность к обобщению. Увеличение числа слоев и нейронов в каждом слое приводит к росту числа параметров модели, что потенциально улучшает ее производительность, но также требует больше вычислительных ресурсов и данных для обучения. Архитектура Allegro++MLP направлена на расширение возможностей оригинальной модели Allegro за счет увеличения ее способности к представлению сложных функций.
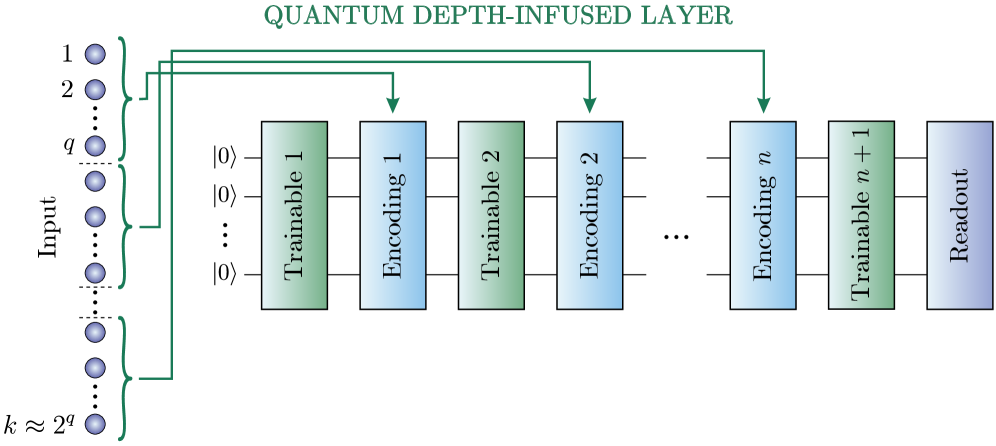
В рамках Allegro++ была внедрена архитектура Allegro++QDI, использующая квантовые слои с углубленной обработкой (QDI) для эффективного управления входными признаками. Данная реализация позволяет оптимизировать обработку данных даже при ограниченном количестве кубитов, что достигается за счет интеграции инструментов, таких как исчисление ZX и ряды Фурье, для оптимизации представления квантовых схем и снижения требований к аппаратным ресурсам. QDI-слои позволяют эффективно кодировать и обрабатывать информацию, минимизируя сложность квантовых вычислений и повышая масштабируемость модели.
Слой QDI оптимизирует представление квантовых схем, используя инструменты, такие как ZX-исчисление и ряды Фурье. ZX-исчисление предоставляет графический язык для манипулирования квантовыми схемами, позволяя упрощать и преобразовывать их без изменения функциональности. Ряды Фурье применяются для эффективного разложения и представления квантовых состояний и операций, что позволяет снизить сложность схем и уменьшить количество необходимых кубитов. Данный подход позволяет добиться более компактного представления квантовых алгоритмов, особенно в условиях ограниченного количества доступных кубитов, и повысить эффективность квантовых вычислений.
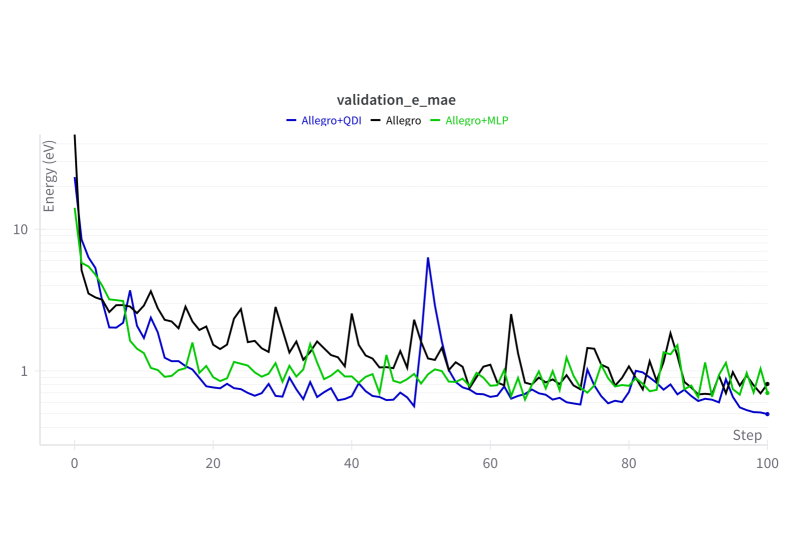
![Обучение всех вариантов Allegro на наборе данных QM9, оптимизированных по гиперпараметрам, демонстрирует сходимость, при этом кривая для Allegro, настроенная согласно [13], соответствует исходным гиперпараметрам QM9, а сглаживание результатов выполнено с использованием экспоненциального скользящего среднего (EMA) с коэффициентом 0.75.](https://arxiv.org/html/2602.16908v1/x8.png)
Строгая Валидация и Оптимизация Гиперпараметров
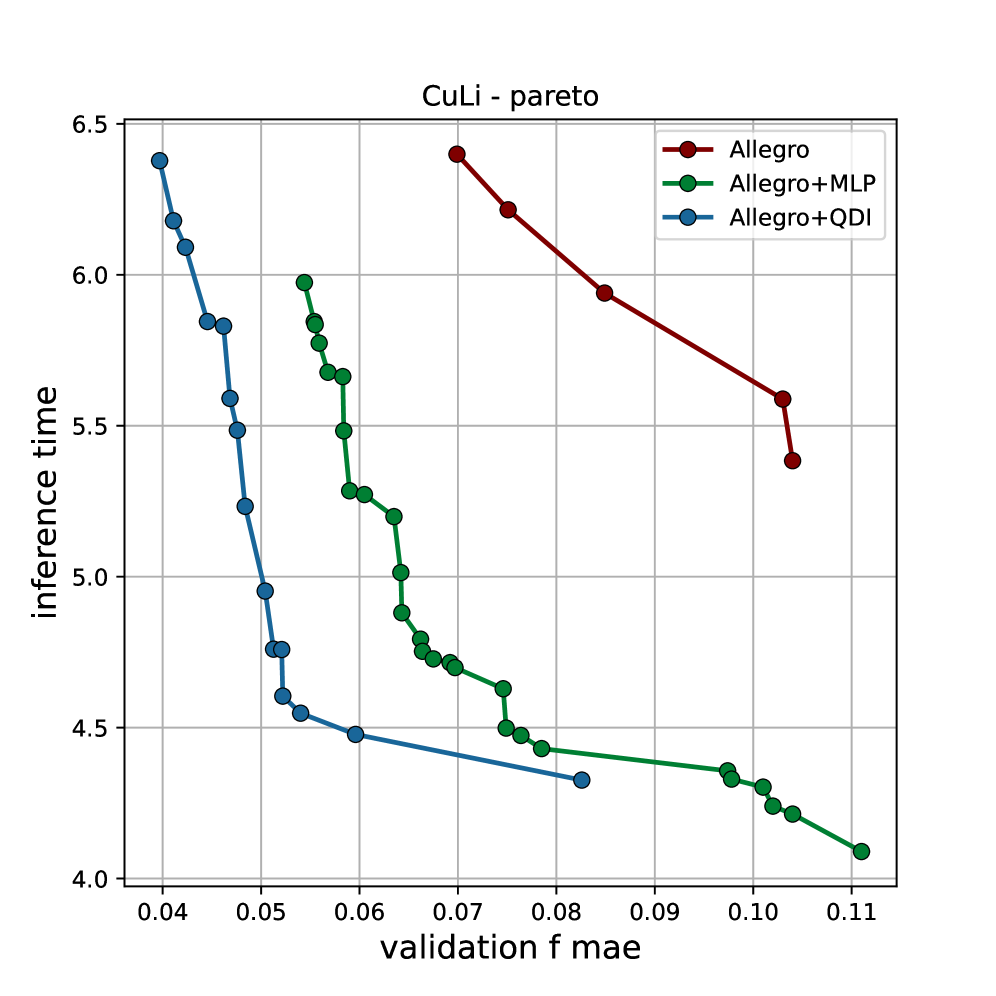
Обучение и валидация моделей проводились на трех различных наборах данных: QM9, rMD17 и специально созданном наборе данных Cu-Li. QM9 представляет собой набор данных, состоящий из молекулярных структур, используемый для оценки производительности моделей в задачах предсказания свойств молекул. rMD17 — это набор данных, содержащий траектории молекулярной динамики, предназначенный для проверки способности моделей к экстраполяции во времени. Набор данных Cu-Li был сгенерирован авторами для оценки производительности моделей в предсказании взаимодействий между литием и медью, что актуально для исследований в области аккумуляторов. Использование разнородных наборов данных позволяет обеспечить широкую применимость разработанных моделей к различным химическим системам и задачам.
Оптимизация гиперпараметров моделей проводилась с использованием алгоритма SAMO-COBRA, который одновременно учитывал два ключевых критерия: точность предсказаний и скорость вычислений (время инференса). SAMO-COBRA является алгоритмом байесовской оптимизации, предназначенным для задач, требующих баланса между производительностью и вычислительными затратами. В процессе оптимизации алгоритм итеративно выбирал комбинации гиперпараметров, оценивал их эффективность на валидационном наборе данных и обновлял модель вероятности, направляя поиск к оптимальным значениям, обеспечивающим наилучшее сочетание точности и скорости работы.
Результаты экспериментов демонстрируют стабильное превосходство вариантов Allegro++ над базовыми моделями по различным метрикам. В частности, при предсказании сил на наборе данных CuLi, модель Allegro++QDI показала увеличение производительности на 13% по сравнению с Allegro++MLP. Данный прирост указывает на эффективность архитектуры QDI в задачах моделирования межмолекулярных взаимодействий и прогнозирования сил, действующих между атомами в исследуемой системе.
К Ускоренному Открытию Материалов: Взгляд в Будущее
Повышенная точность и эффективность моделей Allegro++ значительно ускоряют процессы скрининга и проектирования материалов. Благодаря оптимизации алгоритмов и снижению вычислительных затрат, исследователи получают возможность исследовать более обширные химические пространства, что позволяет идентифицировать новые материалы с заданными свойствами в существенно сокраченные сроки. Это особенно важно для решения сложных задач материаловедения, где традиционные методы часто оказываются слишком трудоемкими и длительными. Возможность быстрого и точного прогнозирования свойств материалов открывает перспективы для создания инновационных технологий в различных областях, от энергетики до электроники и биомедицины.
Снижение вычислительных затрат, обеспечиваемое новыми моделями, открывает беспрецедентные возможности для исследования обширных химических пространств. Это позволяет ученым проводить систематический анализ гораздо большего числа потенциальных материалов, чем было возможно ранее. Благодаря этому, возрастает вероятность обнаружения инновационных соединений, обладающих целевыми свойствами, такими как высокая прочность, проводимость или каталитическая активность. Расширение области поиска, в свою очередь, существенно ускоряет процесс материаловедческих открытий, сокращая время от теоретической разработки до практической реализации и способствуя прогрессу в различных областях науки и техники.
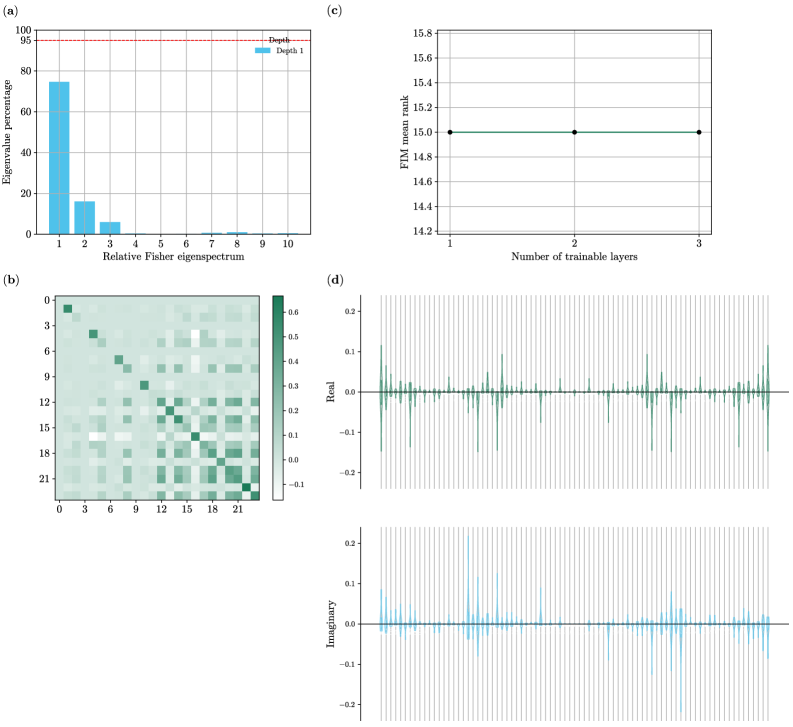
Модель Allegro++QDI демонстрирует высокую выразительность, достигающую 79% благодаря использованию 127 из 161 возможных членов в своем Фурье-спектре. Этот показатель отражает способность модели эффективно описывать сложные энергетические поверхности и предсказывать свойства материалов с высокой точностью. Высокая выразительность достигается за счет оптимального выбора базисных функций, позволяющих адекватно аппроксимировать широкий спектр взаимодействий между атомами. Такое сжатое представление данных, сохраняя при этом значительную выразительность, существенно снижает вычислительные затраты и открывает возможности для эффективного исследования обширных химических пространств в поисках новых материалов с заданными характеристиками.
Данная работа знаменует собой важный шаг на пути к полной реализации потенциала машинного обучения в материаловедении, открывая новую эру ускоренного открытия материалов. Разработанные модели, такие как Allegro++ и Allegro++QDI, не просто повышают точность прогнозирования свойств веществ, но и значительно снижают вычислительные затраты, что позволяет исследователям исследовать гораздо более обширные химические пространства. Это, в свою очередь, создает условия для выявления инновационных материалов с заранее заданными характеристиками, что ранее было затруднительно из-за ограничений в вычислительных ресурсах. Успешная демонстрация высокой экспрессивности модели Allegro++QDI, достигающей 79% при использовании 127 из 161 возможных членов ее Фурье-спектра, подтверждает перспективность данного подхода и предвещает возможность революционных прорывов в области создания новых материалов для различных отраслей науки и техники.
Исследование, представленное в статье, демонстрирует стремление к глубокому пониманию и оптимизации систем машинного обучения для предсказания свойств молекул. Авторы не просто принимают существующие параметры Allegro, а подвергают их тщательному анализу и настройке, используя многоцелевую оптимизацию. Этот подход перекликается с философией Ричарда Фейнмана: «Я не могу объяснить, почему я не могу объяснить это». Подобно тому, как Фейнман стремился докопаться до сути непонятных явлений, авторы статьи стремятся понять, как различные гиперпараметры влияют на точность и эффективность модели, рассматривая даже квантово-классические гибридные варианты для расширения возможностей предсказания энергии и сил в молекулярных системах. Попытка взломать систему, чтобы понять её внутреннюю работу, является ключевой мотивацией данной работы.
Куда Дальше?
Представленная работа, по сути, лишь взлом одной конкретной системы — оптимизации гиперпараметров для Allegro. Однако, за этим взломом проглядывает более глубокая проблема: как создать потенциалы, которые не просто точно предсказывают энергии и силы, но и раскрывают фундаментальные связи между структурой и свойствами вещества. Мультиобъективная оптимизация — это, конечно, шаг вперёд, но она оперирует лишь с заданными критериями. Истинный прорыв потребует переосмысления самих целей оптимизации, возможно, включения в них понятий энтропии, информационной сложности или даже эстетической симметрии.
Гибридные квантово-классические модели выглядят многообещающе, но пока что это, скорее, демонстрация принципиальной возможности, чем реальный выход на качественно новый уровень точности и эффективности. Проблема заключается в том, что квантовые вычисления, несмотря на все обещания, остаются ресурсоёмким и сложным инструментом. Истинный прогресс потребует разработки алгоритмов, которые эффективно используют квантовые преимущества, минимизируя при этом вычислительные издержки.
В конечном итоге, задача состоит не в том, чтобы создать «идеальный» потенциал, а в том, чтобы создать систему, способную к самообучению и адаптации. Представьте себе потенциал, который не просто предсказывает свойства вещества, но и выявляет закономерности, которые ускользают от человеческого понимания. Это и есть та область, где истинный ‘exploit of insight’ ещё предстоит совершить.
Оригинал статьи: https://arxiv.org/pdf/2602.16908.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Улучшение точности квантовых сенсоров: новый подход к подавлению шумов
- Квантовое программирование: Карта развивающегося мира
- Предел возможностей: где большие языковые модели теряют разум?
- Временная запутанность: от хаоса к порядку
- ЭКГ-анализ будущего: От данных к цифровым биомаркерам
- Резонансы в тандеме: Управление светом в микрорезонаторах
- Сердце музыки: открытые модели для создания композиций
- Квантовые кольца: новые горизонты спиновых токов
- Искусственный разум и квантовые данные: новый подход к синтезу табличных данных
- Моделирование спектроскопии электронного пучка: новый подход
2026-02-21 01:32