Автор: Денис Аветисян
Исследователи разработали алгоритм, позволяющий детально изучать структуру тканей и выявлять взаимодействие между клетками на основе анализа пространственной транскриптомики.

Представлен CellScape — фреймворк глубокого обучения, интегрирующий пространственные и геномные данные для точного определения доменов тканей и типов клеток, а также коррекции пакетных эффектов при анализе нескольких образцов.
Несмотря на прогресс в изучении тканевой организации, интеграция пространственного и геномного контекста клеток остается сложной задачей. В работе, озаглавленной ‘Uncovering spatial tissue domains and cell types in spatial omics through cross-scale profiling of cellular and genomic interactions’, представлен CellScape — фреймворк глубокого обучения, совместно моделирующий взаимодействия между клетками в пространстве и геномные связи внутри них. Это позволяет выявлять информативные паттерны, улучшая сегментацию пространственных доменов и анализ разнородных данных пространственной транскриптомики. Какие новые возможности для понимания клеточной организации и выявления ключевых биологических механизмов открывает данный подход?
За гранью усреднения: Ограничения традиционного анализа экспрессии генов
Традиционный анализ экспрессии генов, усредняя сигналы по всему объему ткани, зачастую упускает из виду важнейшие различия в активности генов между отдельными клетками и их микроокружением. Данный подход, хотя и широко распространен, не позволяет выявить пространственную гетерогенность — вариации в экспрессии генов, обусловленные положением клетки в ткани и ее взаимодействием с соседними клетками. В результате, усредненные данные могут не отражать реальную биологическую картину, скрывая тонкие, но значимые различия в фенотипах клеток и их роли в сложных процессах, таких как развитие организма или прогрессирование заболеваний. Игнорирование этих пространственных взаимодействий приводит к упрощенному пониманию биологических систем и ограничивает возможности точной диагностики и разработки эффективных терапевтических стратегий.
Усреднение данных об экспрессии генов в традиционных подходах зачастую скрывает важные детали организации тканей и индивидуальные характеристики клеток. Это приводит к потере ценной информации о взаимодействиях между клетками и их роли в сложных биологических процессах. В результате, понимание механизмов развития, а также патогенеза заболеваний, существенно затрудняется, поскольку не учитывается пространственная неоднородность экспрессии генов и вклад отдельных клеточных фенотипов. Игнорирование этих факторов может привести к неверным интерпретациям и, как следствие, к неэффективным терапевтическим стратегиям.
Для точной интерпретации биологических процессов необходимо учитывать пространственную организацию экспрессии генов в тканях. Традиционные методы, усредняющие сигналы по всей ткани, не позволяют выявить локальные различия в активности генов, которые могут быть критически важны для понимания развития и прогрессирования заболеваний. Изучение экспрессии генов в контексте их точного местоположения внутри ткани открывает возможности для выявления новых клеточных фенотипов и механизмов взаимодействия между клетками, что значительно расширяет наше понимание сложных биологических систем. Без учета пространственного контекста, данные об экспрессии генов могут быть неполными или даже вводящими в заблуждение, препятствуя разработке эффективных терапевтических стратегий.
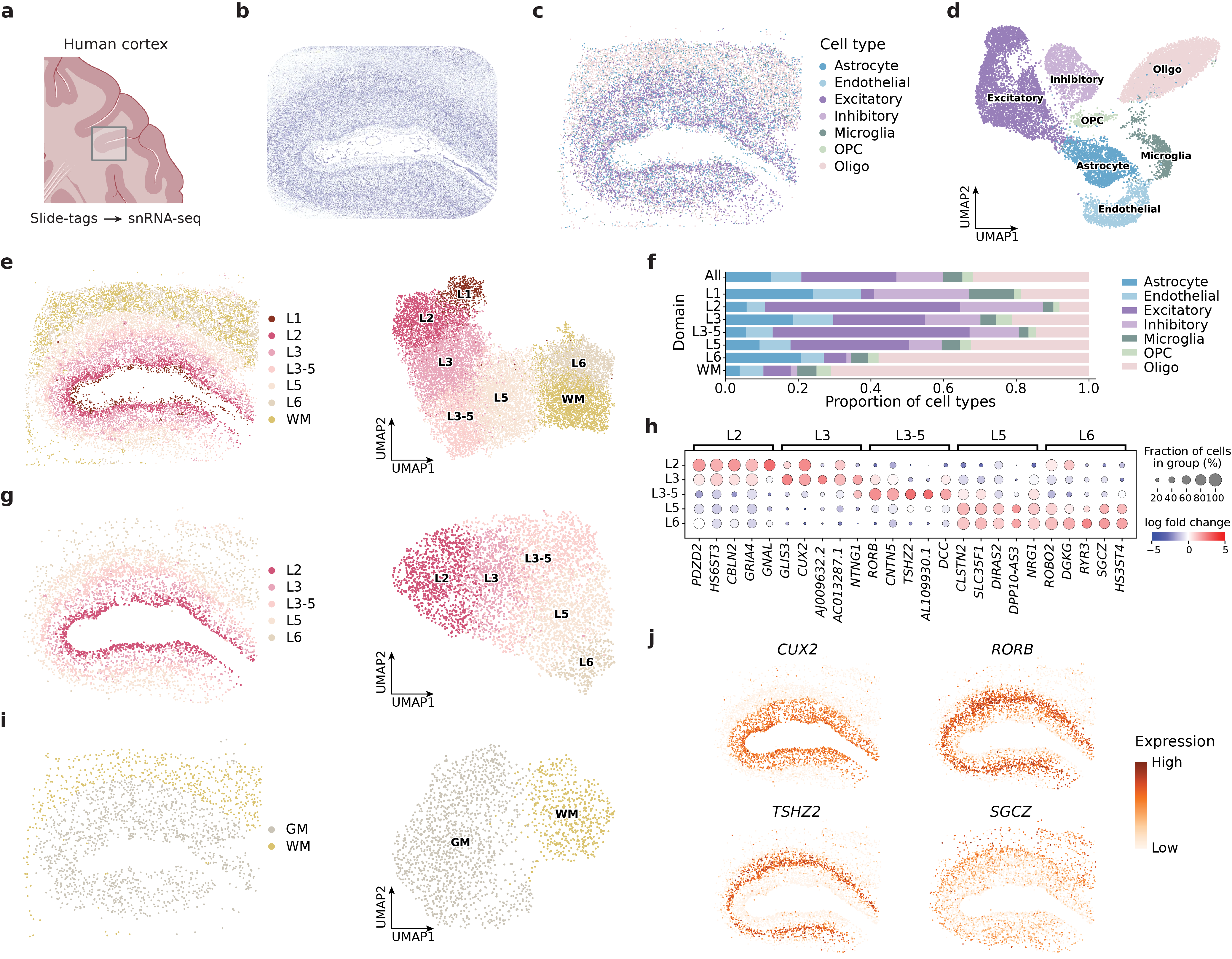
Картирование транскриптома в пространстве: Инструментарий технологий
Пространственная транскриптомика, реализуемая технологиями Visium, Slide-seqV2, STARmap PLUS и Stereo-seq, позволяет исследователям измерять экспрессию генов с сохранением информации о пространственном расположении клеток в ткани. В отличие от традиционных методов секвенирования РНК, которые усредняют сигнал по всей популяции клеток, эти технологии позволяют картировать паттерны экспрессии генов непосредственно в контексте ткани, выявляя пространственные вариации в экспрессии и клеточной организации. Это достигается путем сопоставления транскриптов с их исходным местоположением в ткани, что открывает новые возможности для изучения развития, патогенеза заболеваний и организации тканей.
Технологии пространственной транскриптомики используют различные подходы для определения уровня экспрессии генов с сохранением информации о местоположении. Микрофабрикованные массивы, как в платформе Visium, позволяют проводить анализ на тканях, фиксируя экспрессию в дискретных областях. Методы, основанные на бусинах (bead-based systems), такие как Slide-seqV2, используют пространственно-индексированные олигонуклеотидные бусины для захвата РНК, обеспечивая более высокое разрешение, но требуя более сложной подготовки образцов. Технология STARmap PLUS и Stereo-seq используют ДНК-наношарики (DNA nanoballs) для одновременного измерения экспрессии тысяч генов в каждой пространственной координате, что позволяет достичь субклеточного разрешения, но сопряжено с техническими сложностями и стоимостью.
Различные технологии пространственной транскриптомики, такие как Visium, Slide-seqV2, STARmap PLUS и Stereo-seq, характеризуются компромиссами между разрешением, пропускной способностью и стоимостью. Более высокое разрешение, позволяющее детально изучить экспрессию генов на уровне отдельных клеток, часто достигается за счет снижения пропускной способности — количества анализируемых участков ткани. Методы с высокой пропускной способностью, напротив, могут предоставлять менее детализированную информацию о пространственном распределении экспрессии. Стоимость оборудования и реагентов также значительно варьируется, что требует тщательного выбора технологии, соответствующей конкретным целям исследования и доступным ресурсам. Оптимальный выбор определяется балансом между необходимой степенью детализации, объемом анализируемого материала и бюджетом проекта.
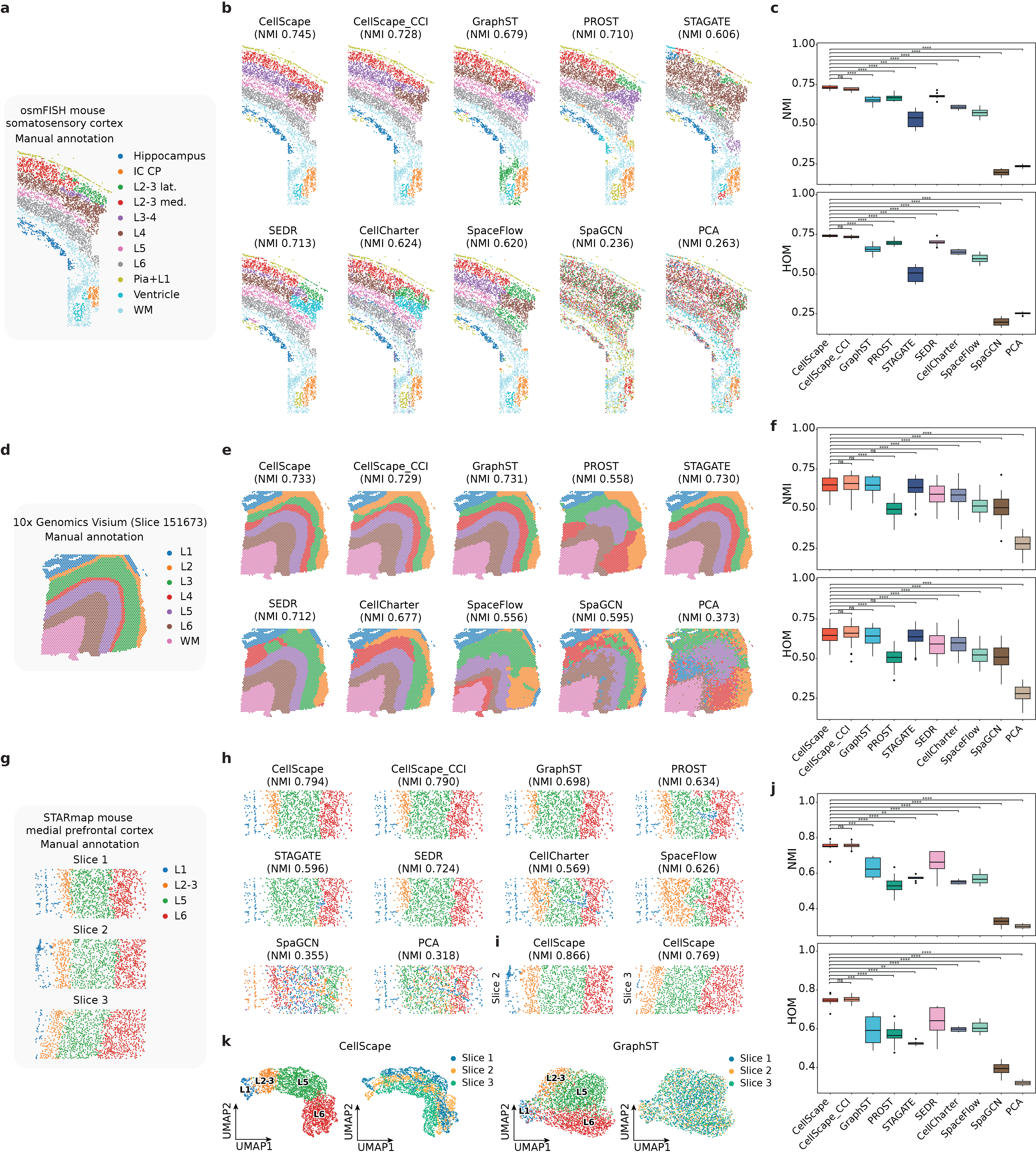
От данных к открытиям: Вычислительный анализ пространственной транскриптомики
CellScape представляет собой платформу глубокого обучения, предназначенную для комплексного анализа организации тканей и фенотипов клеток. Она объединяет данные о пространственном расположении клеток с их геномной информацией, позволяя проводить детальное исследование клеточных взаимодействий и структурной организации тканей. В основе CellScape лежит интеграция методов машинного обучения, включая автоэнкодеры для снижения размерности данных и графовые нейронные сети (GNN) для моделирования взаимосвязей между клетками, что позволяет выявлять сложные паттерны и характеристики тканей на основе как геномных данных, так и пространственной информации.
В CellScape для анализа пространственной транскриптомики применяются методы снижения размерности, такие как Автокодировщики (Autoencoders), позволяющие эффективно представлять данные высокой размерности в более компактном виде. Для моделирования взаимодействия между клетками используются Графовые Нейронные Сети (GNNs), которые рассматривают клетки как узлы графа, а их взаимодействие — как ребра. Такой подход позволяет учитывать пространственную организацию ткани и влияние соседних клеток на экспрессию генов, что повышает точность идентификации клеточных фенотипов и понимания механизмов тканевой организации.
Для оценки качества кластеризации и пространственной когерентности CellScape использует метрики, такие как Нормализованная Взаимная Информация (NMI) и Однородность. На датасете osmFISH, CellScape достиг показателя NMI в 0.745, что является наивысшим результатом среди протестированных методов. NMI измеряет степень согласованности между кластерами, полученными методом, и известными метками клеток, в то время как Однородность оценивает, насколько компактны и четко определены кластеры в пространстве. Высокое значение NMI указывает на эффективную идентификацию и разделение различных типов клеток, а высокая Однородность — на то, что клетки внутри каждого кластера имеют схожие характеристики и пространственное расположение.
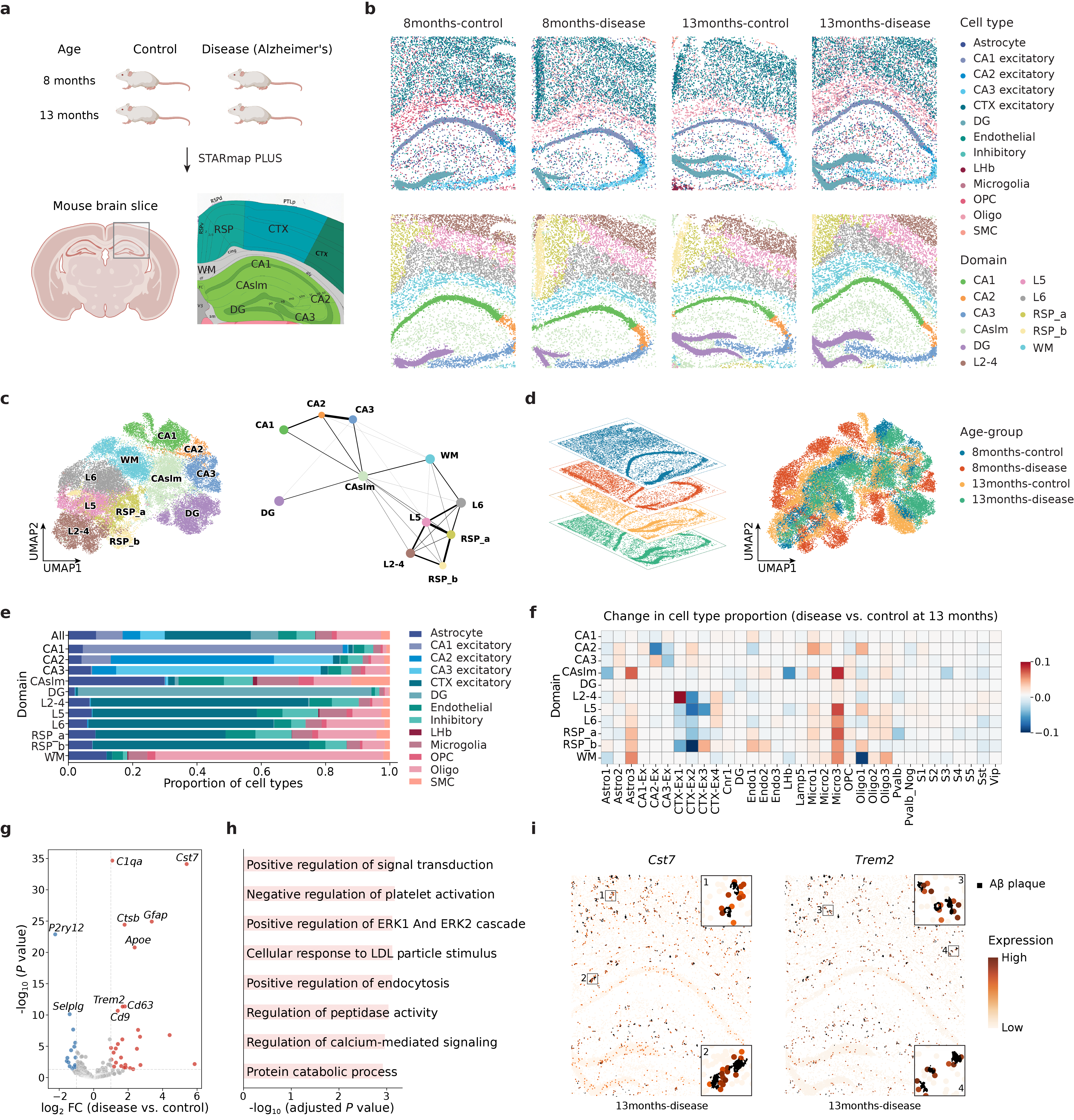
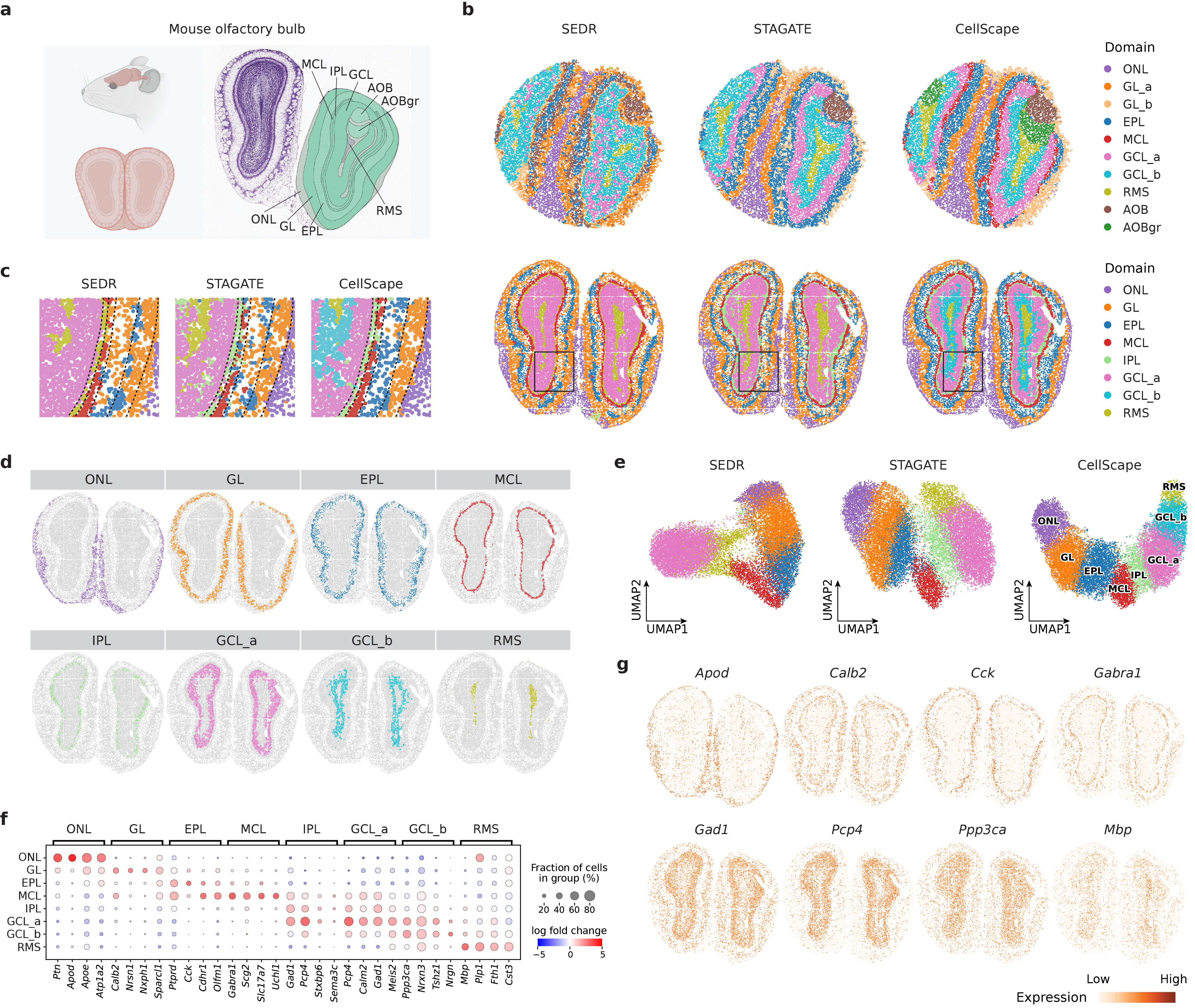
Раскрытие биологических знаний: Применение и перспективы развития
Современные исследования тканей получили мощный инструмент в лице пространственной транскриптомики, особенно в сочетании с алгоритмом CellScape. Данный подход позволяет не просто идентифицировать различные типы клеток, но и точно определить их местоположение внутри ткани, а также выявить сложные взаимодействия между ними. Изучение пространственной организации клеток раскрывает принципы, по которым устроены ткани, позволяя понять, как их структура влияет на функциональность. Благодаря этому, становится возможным более глубокое понимание процессов развития, механизмов заболеваний и эффективности терапевтических вмешательств на уровне отдельных клеток и их окружения. Анализ пространственного распределения генов предоставляет уникальную возможность увидеть, как клетки «общаются» друг с другом и как это влияет на общую работу органа или ткани.
Подход, основанный на пространственной транскриптомике, открывает новые возможности для изучения процессов развития организма, механизмов возникновения заболеваний и эффективности терапевтических вмешательств непосредственно в контексте ткани. Это позволяет исследователям не просто идентифицировать изменения в экспрессии генов, но и точно определить, где в ткани происходят эти изменения, и как они связаны с конкретными клеточными типами и их взаимодействиями. Такая пространственная привязка данных критически важна для понимания сложных биологических процессов, например, формирования органов в эмбриогенезе, прогрессирования опухолей или ответа тканей на лекарственные препараты, предоставляя детальную картину, которую невозможно получить при анализе гомогенизированных образцов.
Разработанный алгоритм CellScape демонстрирует выдающиеся результаты при анализе данных пространственной транскриптомики, последовательно превосходя существующие методы в различных наборах данных. В частности, при анализе набора данных 10x Genomics Visium DLPFC, CellScape достиг показателя однородности в 0.659, что свидетельствует о высокой степени соответствия между предсказанными и эталонными аннотациями. Этот результат подтверждает способность CellScape точно идентифицировать и классифицировать типы клеток на основе их пространственного расположения и экспрессии генов, что делает его ценным инструментом для изучения организации тканей и механизмов развития заболеваний.
Перспективы развития пространной транскриптомики направлены на существенное повышение разрешения, производительности и усовершенствование методов анализа. Ученые стремятся к созданию технологий, способных визуализировать транскриптом на уровне отдельных клеток и даже субклеточных структур, что позволит детально изучить сложные взаимодействия между генами и окружающей средой. Увеличение пропускной способности позволит анализировать более крупные и разнообразные образцы тканей, охватывая большее количество клеток и участков. Совершенствование аналитических методов, включая алгоритмы машинного обучения и статистического моделирования, необходимо для обработки огромных объемов данных и выявления значимых закономерностей. В конечном итоге, эти усовершенствования приведут к всестороннему пониманию транскриптомной организации тканей, открывая новые возможности для изучения развития, болезней и эффективности терапевтических вмешательств.
Исследование, представленное в статье, подобно кропотливому садоводству: для понимания организации тканей необходимо учитывать не только генетические особенности отдельных клеток, но и их пространственное взаимодействие. Авторы предлагают CellScape — инструмент, позволяющий увидеть структуру тканей как единую экосистему, где каждая клетка влияет на соседние. Как однажды заметила Барбара Лисков: «Хороший дизайн состоит из понимания того, что будет изменяться в будущем». Это особенно верно для анализа пространных данных, где архитектура системы должна быть гибкой и способной адаптироваться к новым открытиям и масштабам. CellScape стремится к этой гибкости, интегрируя пространственную и геномную информацию для более точной сегментации и анализа тканей.
Что дальше?
Представленный подход, подобно садовнику, взращивающему ландшафт, а не строящему крепость, выявляет структуру ткани, но не даёт иллюзии полного контроля. Каждая зависимость, выявленная алгоритмом, — это обещание, данное прошлому, гарантия того, что будущие изменения потребуют адаптации. Неизбежно возникнет вопрос: достаточно ли лишь сегментировать домены, или необходимо предсказывать их эволюцию, их ответ на внешние воздействия?
Работа демонстрирует силу интеграции пространственной и геномной информации, но подразумевает, что истинная архитектура ткани — это не статичная схема, а динамичная сеть взаимодействий. Попытки её зафиксировать неизбежно приведут к упрощениям, к потере информации. Всё, что построено, когда-нибудь начнёт само себя чинить — и этот процесс потребует новых инструментов для мониторинга и адаптации моделей.
Очевидно, что следующим шагом станет расширение масштаба — не только в плане объёма данных, но и в плане сложности тканей. Вместо поиска идеальной сегментации, вероятно, стоит сосредоточиться на выявлении механизмов самоорганизации, на понимании того, как отдельные клетки согласованно формируют функциональные структуры. Контроль — это иллюзия, требующая SLA, а истинное знание — это понимание того, как система справляется с неопределенностью.
Оригинал статьи: https://arxiv.org/pdf/2602.12651.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Внимание на границе: почему трансформеры нуждаются в «поглотителях»
- Искусственный нос будущего: как квантовая механика и машинное обучение распознают запахи
- Химический синтез под контролем искусственного интеллекта: новые горизонты
- Внимание в сети: Новый подход к ускорению больших языковых моделей
- Когда большая языковая модель молчит: как избежать галлюцинаций при ответе на вопросы?
- Квантовый скачок в обучении с учителем: новая архитектура для искусственного интеллекта
- Цифровые улики под присмотром ИИ: новая эра криминалистики?
- Обучение представлений для динамических систем: новый взгляд
- Физика под контролем: Как «научить» модели понимать мир
- Симуляция, которая видит себя: новый подход к физическому моделированию
2026-02-16 20:27