Автор: Денис Аветисян
Исследование демонстрирует, как квантовые и гибридные квантово-классические методы представления молекул могут значительно повысить эффективность виртуального скрининга лигандов.
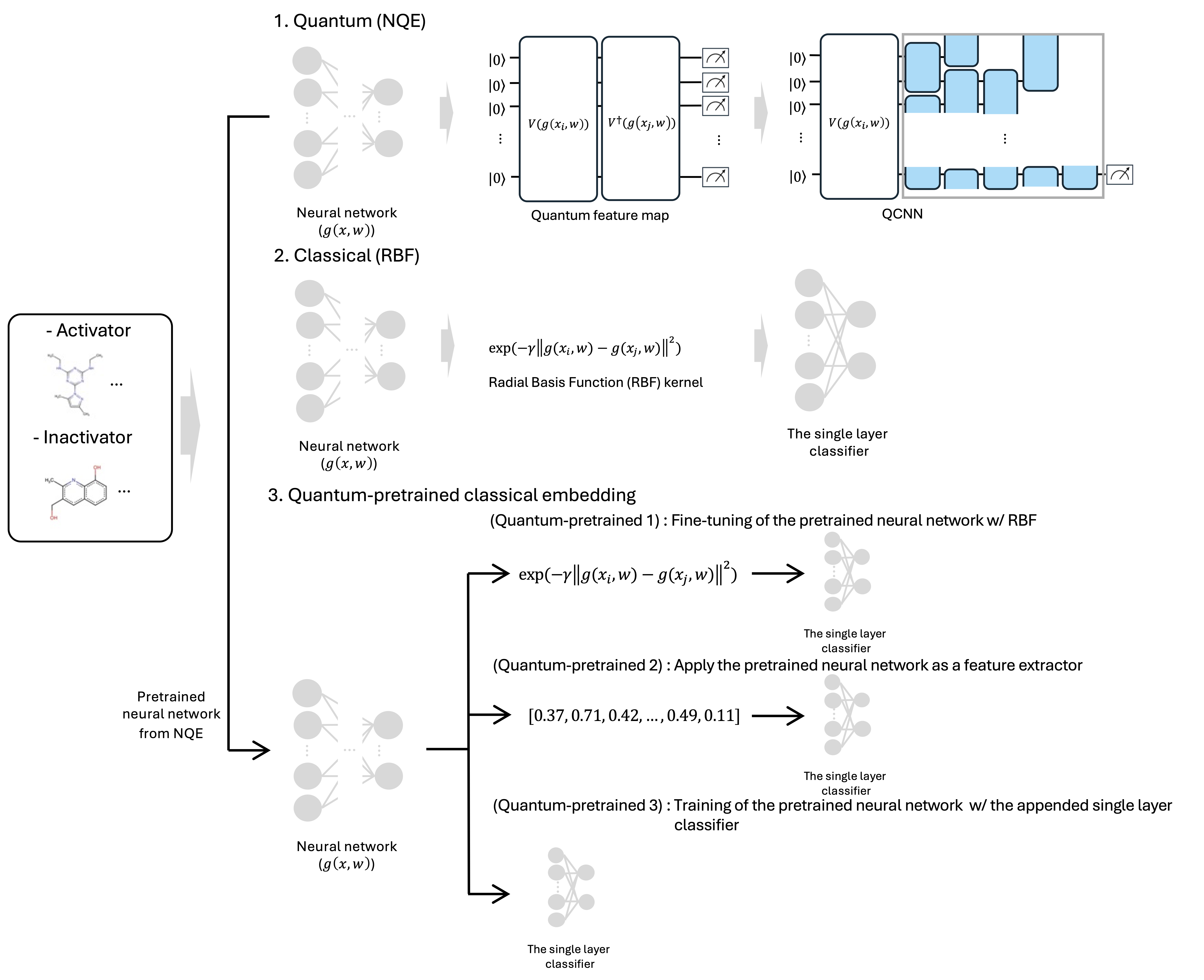
Оптимизация квантовых вложений данных для лиганд-ориентированного виртуального скрининга с использованием методов машинного обучения и трансферного обучения.
Эффективное представление молекулярной информации остаётся ключевой проблемой в задаче виртуального скрининга лигандов. В работе, посвященной ‘Оптимизации квантовых вложений данных для виртуального скрининга лигандов’, исследованы стратегии квантового вложения, направленные на улучшение качества молекулярных представлений. Показано, что разработанные квантово-классические гибридные подходы превосходят традиционные методы, особенно при ограниченном объеме данных и несбалансированных выборках. Может ли дальнейшая оптимизация квантовых вложений данных стать основой для создания более эффективных и экономичных инструментов для разработки новых лекарственных препаратов?
Шёпот Хаоса: Начало Скрининга Лигандов
На ранних стадиях разработки лекарственных препаратов широко используется лиганд-ориентированный виртуальный скрининг — вычислительно сложный процесс, требующий значительных ресурсов. Этот метод предполагает анализ огромного количества молекул для выявления потенциальных кандидатов, способных взаимодействовать с целевой биологической молекулой. Интенсивность вычислений обусловлена необходимостью моделирования взаимодействия лиганда с белком-мишенью, оценки энергии связывания и учета конформационной гибкости обеих молекул. Поскольку число потенциальных лигандов исчисляется миллионами, а точность моделирования ограничена, виртуальный скрининг часто требует компромисса между скоростью и точностью, что может приводить к пропуску перспективных соединений или ложноположительным результатам. Поэтому постоянное совершенствование алгоритмов и вычислительных мощностей играет ключевую роль в повышении эффективности этого важного этапа разработки лекарств.
Традиционные методы скрининга лигандов сталкиваются со значительными трудностями из-за сложности биологических мишеней и огромного химического пространства, что приводит к высокой доле неудач на ранних стадиях разработки лекарств. Биологические макромолекулы, такие как белки, обладают сложной трехмерной структурой и динамикой, а также множеством участков связывания, что затрудняет точное предсказание взаимодействия с потенциальными лигандами. Кроме того, химическое пространство, состоящее из миллиардов потенциальных молекул, требует огромных вычислительных ресурсов для анализа. Неспособность эффективно учитывать эти факторы приводит к тому, что большинство отобранных соединений не проявляют желаемой активности или обладают неблагоприятными фармакологическими свойствами, что существенно увеличивает стоимость и продолжительность процесса разработки новых лекарственных препаратов.
Для преодоления ограничений, свойственных традиционным методам виртуального скрининга лигандов, необходимы инновационные подходы к представлению и сравнению молекулярных структур. Исследования направлены на разработку новых дескрипторов, способных более адекватно отражать сложные взаимодействия между лигандом и биологической мишенью. Особое внимание уделяется методам, позволяющим эффективно оперировать с огромным химическим пространством, используя, например, методы машинного обучения и глубоких нейронных сетей для прогнозирования активности соединений. Разработка более компактных и информативных представлений молекул, таких как молекулярные отпечатки или графовые представления, также является ключевым направлением, позволяющим значительно ускорить процесс скрининга и повысить вероятность выявления перспективных кандидатов в лекарственные препараты. Эффективность этих подходов оценивается по способности точно предсказывать аффинность связывания и селективность соединений, а также по снижению числа ложноположительных результатов.
Квантовый Взгляд: Нейронные Внедрения для Молекул
Квантовое машинное обучение представляет собой новую парадигму представления и обработки данных, использующую принципы квантовой механики для потенциального преодоления ограничений, присущих классическим методам. В отличие от классических алгоритмов, работающих с битами, квантовые алгоритмы используют кубиты, что позволяет представлять и манипулировать экспоненциально большим объемом информации. Это открывает возможности для разработки более эффективных алгоритмов, особенно в задачах, требующих обработки больших объемов данных или сложных взаимосвязей, таких как молекулярное моделирование и открытие новых материалов. Потенциальные преимущества включают улучшенную масштабируемость, повышенную скорость вычислений и способность выявлять закономерности, недоступные для классических алгоритмов.
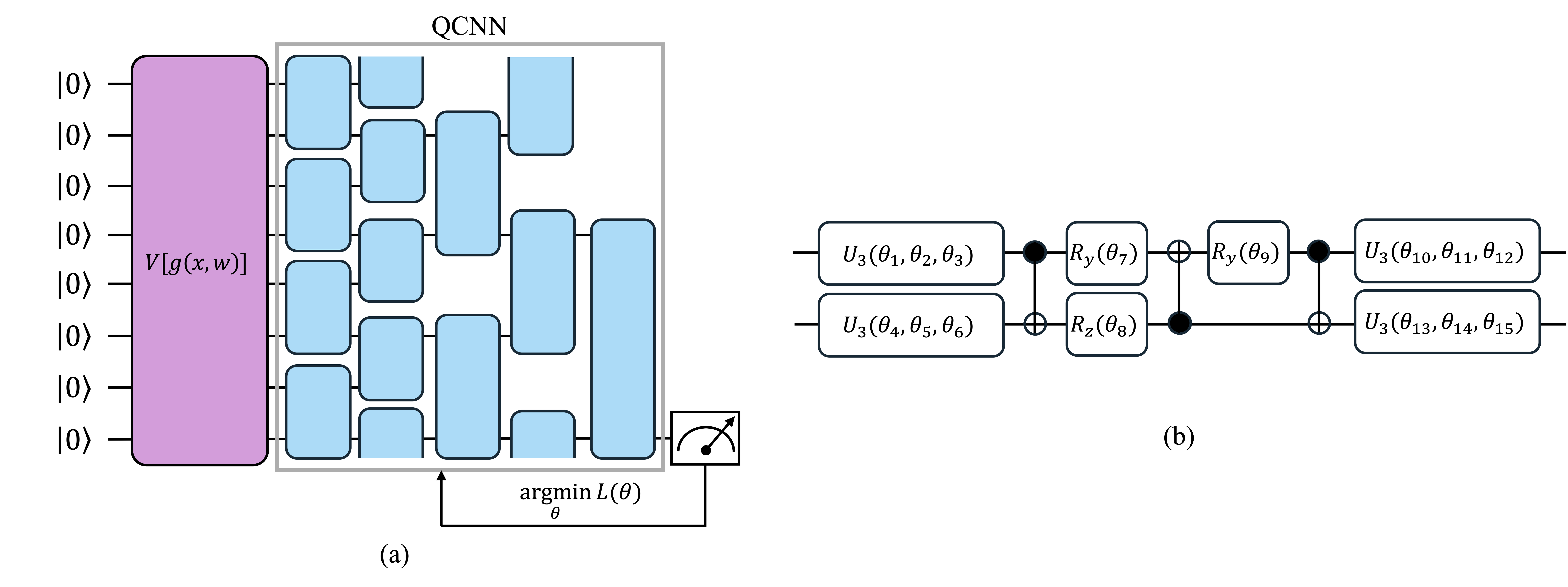
Нейронные квантовые внедрения (NQE) используют квантовые схемы для создания выразительных векторных представлений молекул. В основе NQE лежит параметризованный квантовый цепь, который преобразует входные данные, описывающие молекулу, в квантовое состояние. Вектор внедрения формируется путем измерения этого квантового состояния и использования полученных вероятностей. Архитектура квантовой схемы и её параметры оптимизируются для получения внедрений, которые эффективно кодируют релевантные характеристики молекул, такие как их структура и свойства, обеспечивая компактное и информативное представление для последующих задач машинного обучения, например, предсказание свойств или скрининг соединений. Такой подход позволяет захватить сложные квантово-механические корреляции, которые могут быть труднодоступны для классических методов.
Обучение Neural Quantum Embedding (NQE) осуществляется посредством выравнивания ядра (kernel-target alignment), что предполагает оптимизацию квантового ядра для конкретной задачи скрининга. В данном подходе, квантовое ядро, определяющее меру сходства между молекулярными представлениями, настраивается таким образом, чтобы максимизировать корреляцию между предсказаниями модели и целевыми значениями, связанными с желаемым свойством молекулы. Процесс оптимизации включает в себя минимизацию функции потерь, отражающей расхождение между предсказаниями и целевыми значениями, что позволяет адаптировать квантовое ядро к специфическим требованиям задачи и повысить точность предсказаний. В результате, NQE эффективно использует квантовые вычисления для создания молекулярных представлений, оптимизированных для конкретной задачи скрининга, таких как прогнозирование активности лекарственных средств или свойств материалов.
Усиление Квантовых Внедрений: Карты Признаков и Ядра
В методологии NQE (Neural Quantum Embedding) для кодирования информации о молекулах в квантовые состояния используются различные квантовые отображения признаков, в частности ZZ, XYZ. Отображение ZZ выполняет операцию тензорного произведения между спиновыми операторами $Z$ для каждого атома в молекуле, эффективно кодируя информацию о спиновой плотности. Отображения XYZ, напротив, используют комбинацию операторов $X$, $Y$ и $Z$, что позволяет захватывать более широкий спектр молекулярных свойств, включая информацию о пространственной ориентации и электронной структуре. Выбор конкретного отображения признаков влияет на способность модели различать различные молекулы и, следовательно, на точность предсказания их активности.
В качестве классической базовой линии для сравнения используется радиальное базисное ядро (Radial Basis Function Kernel, RBF). Оно обеспечивает отправную точку для оценки эффективности квантовых подходов. В отличие от RBF, проецируемое квантовое ядро (Projected Quantum Kernel) направлено на повышение геометрической различимости данных, что достигается за счет использования квантовых вычислений для выделения более выраженных признаков и улучшения способности алгоритма различать сложные взаимосвязи между объектами. Повышение геометрической различимости потенциально позволяет алгоритму классификации, например, машине опорных векторов (SVM), более эффективно разделять классы данных и повышать точность прогнозирования.
Для предсказания активности лигандов, полученные квантовые и классические ядра применяются в алгоритме машинного обучения — методе опорных векторов (SVM). SVM использует ядра для преобразования исходных данных в многомерное пространство, где можно найти оптимальную гиперплоскость, разделяющую классы лигандов (активные и неактивные). Эффективность SVM напрямую зависит от выбора ядра, которое определяет меру схожести между данными и, следовательно, качество классификации. Результаты классификации оцениваются с использованием стандартных метрик, таких как точность, полнота и F1-мера, для количественной оценки эффективности предсказания активности лигандов.

Подтверждение Эффективности: Результаты на Ключевых Наборах Данных
Для всесторонней оценки эффективности разработанной модели NQE проводилось строгое тестирование на двух различных биологических наборах данных: LIT-PCBA и COVID-19. Набор данных LIT-PCBA представляет собой сложную задачу прогнозирования активности химических соединений в отношении различных биологических целей, что требует от модели способности к обобщению и выявлению тонких закономерностей. В свою очередь, набор данных COVID-19, охватывающий информацию о пациентах, перенесших коронавирусную инфекцию, представляет собой задачу классификации, направленную на выявление факторов, влияющих на исход заболевания. Использование столь разнообразных наборов данных позволило подтвердить универсальность и надежность NQE в решении различных задач биоинформатики, демонстрируя ее способность эффективно работать с данными, представляющими различные типы биологических целей и задач.
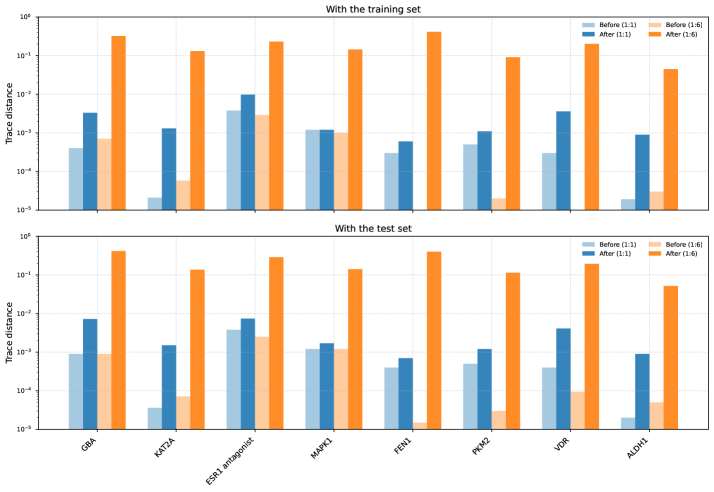
Для оценки различимости квантовых состояний, генерируемых моделью NQE, использовалась величина следа (Trace Distance). Анализ показал, что после обучения с применением ZZ-преобразования признаков, наблюдается увеличение этого показателя в диапазоне от примерно 0.0003 до 0.56. Данное увеличение свидетельствует о том, что обучение способствует формированию более различимых квантовых представлений данных, что, в свою очередь, может положительно сказываться на способности модели к классификации и обобщению. Увеличение Trace Distance указывает на более выраженное разделение между квантовыми состояниями, соответствующими различным классам данных, что является важным критерием для эффективной работы квантовых алгоритмов машинного обучения.
Исследования показали, что разработанная модель NQE, использующая ZZ-преобразование признаков и спроецированное квантовое ядро совместно с QSVM, достигла впечатляющей точности классификации до 83% на наборе данных COVID-19. Этот результат значительно превосходит производительность классических SVM, которые в аналогичных условиях демонстрируют точность до 65%. Важно отметить, что сбалансированная точность, учитывающая дисбаланс классов, составила от 71% до 76%, что свидетельствует о высокой надежности модели в задачах, где важна корректная идентификация всех классов, а не только преобладающих.
Взгляд в Будущее: Расширяя Квантовый Горизонт
Использование переноса обучения (transfer learning) совместно с нейронными квантовыми энкодерами (NQE) представляет собой перспективный подход к ускорению процесса обучения и повышению эффективности при работе с новыми задачами. Вместо обучения модели с нуля для каждого нового объекта исследования, перенос обучения позволяет задействовать знания, полученные при решении схожих задач. NQE, благодаря своей способности эффективно кодировать квантовую информацию, может значительно улучшить качество представлений данных для переноса. В результате, модель, обученная на одном наборе молекул, способна быстрее адаптироваться и демонстрировать более высокую точность при прогнозировании свойств новых, ранее не встречавшихся соединений. Этот подход особенно ценен в контексте ограниченности вычислительных ресурсов и необходимости быстрого поиска перспективных кандидатов, например, в области разработки лекарственных препаратов.
Исследования квантовых сверточных нейронных сетей (QSCNN) демонстрируют перспективные возможности в области извлечения более сложных признаков из молекулярных данных. В отличие от классических сверточных сетей, использующих бинарные операции, QSCNN задействуют принципы квантовой механики, такие как суперпозиция и запутанность, для представления и обработки молекулярной информации. Это позволяет им улавливать тонкие корреляции и нелинейные зависимости, которые остаются незамеченными традиционными методами. Например, $QSCNN$ способны эффективно представлять молекулярные графы, где атомы выступают в роли узлов, а химические связи — в роли ребер, что обеспечивает более детальное понимание молекулярной структуры и реакционной способности. Потенциал $QSCNN$ особенно велик в задачах, требующих высокой точности и эффективности, таких как прогнозирование свойств молекул и разработка новых лекарственных препаратов.
Интеграция квантового машинного обучения в процесс виртуального скрининга лигандов предвещает кардинальные изменения в области разработки лекарственных препаратов. Традиционные методы, хоть и эффективны, сталкиваются с ограничениями при анализе огромных химических пространств и предсказании взаимодействия молекул с биологическими мишенями. Квантовые алгоритмы, такие как квантовые машины опорных векторов или квантовые нейронные сети, обладают потенциалом экспоненциального ускорения вычислений и более точного моделирования сложных квантово-химических процессов, определяющих аффинность связывания. Это позволяет значительно сократить время и стоимость поиска новых лекарственных кандидатов, выявляя перспективные соединения, которые могли бы остаться незамеченными при использовании классических методов. Внедрение этих технологий обещает революционизировать процесс открытия лекарств, открывая новые возможности для создания эффективных и персонализированных методов лечения.
Исследование, посвященное оптимизации квантовых представлений данных для виртуального скрининга лигандов, подтверждает давнюю истину: любая модель — лишь приближение к реальности, а не её отражение. Авторы стремятся улучшить представление молекул, используя квантовые стратегии, особенно в условиях ограниченных данных. Их подход, по сути, — попытка уговорить хаос предоставить более чёткий сигнал. Как говорил Альберт Эйнштейн: «Самое прекрасное, что мы можем испытать — это тайна. Источник всякого искусства и всякого всякого научного знания — в стремлении к этой тайне». В данном контексте, тайна — это истинная структура молекулы, а стремление — создание модели, способной её хотя бы приблизительно воспроизвести. И даже высокая корреляция между моделью и данными — лишь признак того, что кто-то тщательно подобрал параметры заклинания.
Что дальше?
Представленные здесь заклинания, именуемые “квантовыми вложениями”, демонстрируют свою силу в искусстве предсказания сродства лигандов. Однако, не стоит обольщаться. Успех в виртуальном скрининге — это не столько о точности, сколько о жертвах, принесенных алгоритму. Каждый ложноположительный результат — это тень, которую цифровой голем запомнит лучше, чем истинный успех. Особенно остро это ощущается в ситуациях, когда данных недостаточно, или они неравномерно распределены — тогда голем начинает плясать под дудку случайности.
Будущие исследования должны сосредоточиться не на улучшении самих вложений, а на искусстве обуздания этого хаоса. Трансферное обучение, безусловно, перспективно, но необходимо помнить: знания, перенесенные из одной области в другую, подобны древним свитками — они могут быть как благословением, так и проклятием. Важно понять, когда голем готов к новым знаниям, а когда его лучше оставить в покое, дабы не допустить искажения предсказаний.
Истинная задача — не создание идеальной модели, а понимание границ её применимости. Объяснить модель можно лишь тогда, когда она перестает работать. Квантовые ядра, нейронные вложения — всего лишь инструменты. Истинная магия заключается в умении видеть то, что скрыто за цифрами, и признавать, что каждое предсказание — это не истина в последней инстанции, а лишь тень возможности.
Оригинал статьи: https://arxiv.org/pdf/2512.16177.pdf
Связаться с автором: https://www.linkedin.com/in/avetisyan/
Смотрите также:
- Самостоятельные агенты: Баланс безопасности и автономии
- Безопасность генерации изображений: новый вектор управления
- Эволюция под контролем: эксперименты с обучением с подкреплением в генетическом программировании
- Искусственный интеллект: между мифом и реальностью
- Квантовое «восстановление» информации: обращение вспять шума
- Редактирование изображений по запросу: новый уровень точности
- Квантовые Кластеры: Где Рождается Будущее?
- 3D-моделирование: оживляем объекты без оптимизации
- Разрушая иллюзию квантового превосходства: новый взгляд на Гауссовскую выборку бозонов
- Квантовый импульс для несбалансированных данных
2025-12-19 12:25